Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Los datos financieros del órgano encargado de autorizar el uso de fármacos en la UE, a los que ha tenido acceso Investigate Europe, muestran su dependencia de multinacionales como Novartis, Pfizer o AstraZeneca.
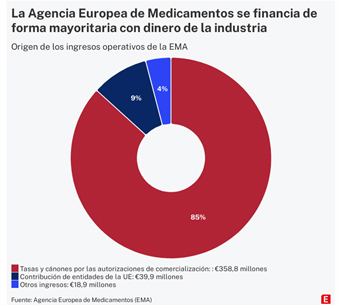
La Agencia Europea de Medicamentos (EMA por sus siglas en inglés) es responsable de autorizar la comercialización de los fármacos que se consumen en España y en el resto de la UE contra las principales enfermedades. Sus decisiones impactan directamente en la cuenta de resultados de las empresas del sector. Al mismo tiempo, un reducido grupo de 21 multinacionales farmacéuticas aportan casi la mitad de los más de 400 millones que ingresa anualmente el órgano regulador.
Investigate Europe desvela, por primera vez, quiénes son las compañías que financian a la EMA. Los periodistas consiguieron los datos a través de una solicitud de acceso a información pública al amparo de la legislación europea sobre Transparencia. En concreto, pidieron conocer la identidad y el importe abonado por todas las empresas farmacéuticas en 2022. La EMA aportó un archivo con los 41.640 pagos recibidos ese año, realizados por 3.564 entidades diferentes. La Agencia cobra tasas por las solicitudes de autorización de venta de fármacos y por los cambios que se producen en las mismas, así como tasas anuales por los medicamentos autorizados.
El análisis de los datos permite alcanzar una conclusión principal: aunque son miles las entidades que abonan tasas, un reducido grupo de 21 multinacionales acapara la mitad de los pagos. En otras palabras: el presupuesto de la EMA depende en buena medida de un pequeño número de empresas, que son a su vez las principales beneficiadas por las decisiones que toma.
Novartis encabeza la lista de pagos de 2022, con €19,6 millones, seguida por Pfizer (14,3), AstraZeneca (12,5), Janssen (10,5), Roche (10,2) y GlaxoSmithKline (10,2). En total, las 21 multinacionales aportaron €165,4 millones (ver el listado completo en el gráfico que aparece en el artículo original – enlace que aparece en el encabezado).
Aunque la EMA aportó el detalle de todos los ingresos recibidos, al entender que estaba obligada a ello por la legislación sobre Transparencia, censuró la identidad de algunos pagadores. Así lo explicó en una carta enviada a los periodistas: “Se ha suprimido la información comercial confidencial relacionada con futuros planes de desarrollo. En particular, se han suprimido los nombres de los terceros que han pagado honorarios por el asesoramiento científico para evitar que la divulgación del documento menoscabe la protección de los intereses comerciales de una persona física o jurídica, incluida la propiedad intelectual”.
Además de las tasas que recibe por las autorizaciones de comercialización, la Agencia también cobra a las empresas que lo solicitan por darles asesoramiento científico mientras están desarrollando un producto médico.
La información censurada afecta a pagos por importe de €36,2 millones, lo que equivale al 10% de los 364,2 millones que la EMA ingresó de empresas farmacéuticas en 2022. Por tanto, la cantidad total abonada por las 21 multinacionales es aún mayor, ya que son ellas las que tienen más fármacos en desarrollo con asesoramiento científico de la Agencia.
La dependencia que tiene la EMA del dinero de la industria es casi total y se ha incrementado de forma incesante durante los últimos años.
Cuando se creó en 1995, sólo el 20% de la financiación procedía de las empresas. El resto salía de los presupuestos de la Unión Europea. En 2022, los ingresos de explotación de la Agencia ascendieron a 417 millones, de forma que los pagos de las farmacéuticas supusieron más del 85% del total [en las cuentas anuales figuran ingresos por 358,8 millones como se ve en el gráfico, mientras que en la información facilitada a través de Transparencia son 364,2, porque esta cifra incluye 5,4 millones ingresados en 2022 procedentes de años anteriores]. En 2024 se prevé que más del 90% proceda de las tasas que abona la industria. La aportación de la UE se ha ido reduciendo en paralelo y en 2022 quedó por debajo del 10% de los ingresos.
El ascenso de los ingresos procedentes de las farmacéuticas se debe en parte a que los procesos de autorización se han disparado. El regulador europeo ha dado luz verde a una media de 85 nuevos medicamentos al año en la última década, frente a una media anual de 49 en los 15 años anteriores.
Hay que señalar que este modelo, por el que las empresas farmacéuticas financian al órgano que las regula, es el que se aplica también en las agencias estatales de la mayoría de los países europeos. Con una gran excepción: Francia. El 92% de los ingresos de la Agencia Nacional para la Seguridad de los Medicamentos gala proceden de la Seguridad Social, a través del seguro de enfermedad (assurance maladie). Tienen, por tanto, origen público.
Por su parte, la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) tuvo en 2022 unos ingresos de €83,2 millones, de los que 68,9 fueron tasas pagadas por la industria, lo que equivale al 82,8% del total. Otros 11,7 millones (14,0% del total) fueron ingresos por servicios prestados a la EMA correspondientes a las actividades de autorización y seguimiento de los fármacos aprobados por el procedimiento centralizado.
La venta de un medicamento en un país de la UE puede ser autorizada por tres vías: por la EMA, en lo que se conoce como procedimiento centralizado; por las Agencias estatales como la AEMPS, o a través del reconocimiento mutuo y del procedimiento descentralizado, por el que un país aprueba un fármaco ya autorizado en otro Estado. De los 1.800 nuevos permisos anuales de medicamentos en España, el 12% recibe autorización de la EMA, el 33% de la AEMPS y el 55% sigue procedimientos descentralizados o de reconocimiento mutuo.
Sin embargo, la EMA tiene el monopolio para aprobar todos los medicamentos contra el cáncer, la diabetes, las dolencias neurodegenerativas, las enfermedades raras o virales y los producidos a través de procesos biotecnológicos o terapias genéticas. Por eso, aunque en términos cuantitativos sean menos los fármacos que aprueba la EMA, en términos cualitativos su importancia es mucho mayor. Y por supuesto también en términos económicos para las empresas, que generan la inmensa mayoría de sus beneficios gracias a fármacos aprobados por la EMA.
Para realizar los procesos de evaluación, la EMA recurre a investigadores de las Agencias estatales, y eso es lo que explica que el 14% de los ingresos de la española AEMPS procedan de la Agencia europea.
Este modelo de financiación tanto de la EMA como de la mayoría de las Agencias estatales despierta críticas entre expertos consultados por Investigate Europe ante los potenciales conflictos de interés. Los estrechos vínculos de la EMA con la industria son de sobra conocidos, afirma Yannis Natsis, quien formó parte de su consejo durante dos años y medio, en representación de los proveedores sanitarios. “La EMA tiene una larga tradición de estrecha colaboración con las empresas que se supone que debe regular”, indica Natsis.
El médico Fernando Lamata, uno de los principales expertos españoles en el tema del precio de los medicamentos, considera que “no debería haber financiación privada” ni de la EMA ni de las Agencias estatales. La razón para él es obvia: “En el sistema actual, ¿quién va a analizar los datos de los ensayos clínicos? La EMA. ¿Y quién paga a la EMA? La misma industria que realiza los estudios que tiene que analizar la EMA”.
En la Agencia Europea de Medicamentos lo ven de otra forma. Una portavoz de este organismo recuerda que las autoridades europeas ya tomaron a mediados de los noventa “la decisión política de exigir a las empresas farmacéuticas que contribuyeran a sufragar los costes de la regulación y las evaluaciones de medicamentos realizadas por la EMA”. En su opinión, es una cuestión de justicia con los contribuyentes: “Parece justo, teniendo en cuenta que las autorizaciones de comercialización aportan considerables ventajas económicas al solicitante (es decir, acceso al mercado único de la UE), que el coste de la evaluación científica y el seguimiento posterior a la autorización de los medicamentos sea compartido por el solicitante y no corra exclusivamente a cargo de los contribuyentes”. De lo contrario, añade esta portavoz, “las empresas se beneficiarían doblemente, es decir, primero por acceder al mercado de la UE, donde pueden obtener beneficios, y segundo por no pagar ningún coste reglamentario por acceder a él”.
Además, la EMA niega que los pagos de la industria afecten a las decisiones que adopta: “Los solicitantes pagan por un procedimiento, pero no por el resultado del mismo. Esto significa que una empresa paga en el momento de presentar una solicitud a la EMA; a continuación, la Agencia lleva a cabo una evaluación independiente. Utilizando la analogía de un examen de conducir, hay que pagar para hacer un examen de conducir, pero no hay garantía de aprobar el examen”.
Farmaindustria, la patronal española, tampoco ve ningún problema en que sus empresas paguen a la Agencia que las regula: “La Ley establece unas tasas para las compañías farmacéuticas por los servicios que presta la AEMPS, como sucede con muchos servicios públicos en los que el usuario paga tasas precisamente por el uso de esos servicios. Este pago de tasas no genera ningún tipo de conflicto de interés, como es obvio”. En todo caso, añade su portavoz, “la industria no tendría ningún inconveniente en que se suprimieran las tasas de la AEMPS, que no dejan de ser un coste adicional para las empresas”.
En parte de la comunidad científica preocupa que la EMA esté autorizando el uso de fármacos con ensayos clínicos poco exigentes sobre su eficacia y seguridad. “La EMA está aprobando nuevos medicamentos con mayor rapidez y con menos datos clínicos disponibles. Cada vez nos resulta más difícil evaluar su beneficio añadido real en comparación con los fármacos existentes”, afirma Beate Wiseler, del prestigioso instituto IQWiG, responsable de la calidad y eficiencia sanitarias en Alemania.
Esa preocupación se dispara entre los expertos al analizar los procedimientos especiales que tiene la EMA para garantizar un acceso rápido al mercado de determinados fármacos. Existen tres vías aceleradas para obtener el permiso: la autorización condicional de comercialización (CMA, por sus siglas en inglés), la evaluación acelerada y las circunstancias excepcionales.
Investigate Europe ha analizado todos los fármacos aprobados por la EMA desde 2004, cuando se introdujo el primero de estos procedimientos especiales, hasta diciembre de 2023. En total, 198 medicamentos accedieron al mercado a través de alguna de las tres vías aceleradas. De ellos, 173 se siguen comercializando, 16 fueron retirados por los laboratorios, en 7 casos expiró la licencia y sólo en dos ocasiones la Agencia revocó la autorización (Lartruvo, patentado por Eli Lilly y usado para el tratamiento de determinados sarcomas, y Adakveo, propiedad de Novartis y autorizado para prevenir crisis vaso-oclusivas).
Una portavoz de la EMA destacó que desde 2004 la Agencia ha autorizado “más de 1.400 medicamentos”, por lo que los aprobados por una de las vías aceleradas representan el 14%.
La vía más utilizada, y la que genera mayor atención crítica de la comunidad científica, es la autorización condicional de comercialización. Esta se otorga con información insuficiente sobre la eficacia o seguridad del medicamento, para hacer frente a “necesidades médicas insatisfechas”, a condición de que la empresa aporte con posterioridad la evidencia científica que falta. Si lo hace, el medicamento obtiene una autorización estándar.
El análisis de Investigate Europe ha descubierto tres hechos llamativos: que el recurso a este procedimiento se ha multiplicado en los últimos cinco años; que dos tercios de los fármacos aprobados por esta vía son propiedad del grupo de 21 multinacionales que acaparan la financiación de la EMA, y que los laboratorios pueden llegar a tardar hasta diez años en presentar la evidencia científica que falta.
La Agencia concedió la primera autorización condicional en 2006 y hasta finales de 2023 la otorgó en total a 91 medicamentos. Pues bien, 51 de ellos recibieron el visto bueno en los últimos cinco años, mientras que en los trece años anteriores sólo lo obtuvieron 40. Al ser preguntada por estos datos, la portavoz de la EMA destacó que el recurso a este procedimiento para aprobar vacunas contra la covid-19 podía explicar en parte esos números. Sin embargo, sólo siete medicamentos relacionados con la Covid-19 recibieron este tipo de autorización. Por tanto, aun sin tener estos siete fármacos en cuenta, la conclusión es la misma: entre 2019 y 2023 la Agencia utilizó esta vía especial en más ocasiones que en los trece años anteriores.
Los datos también demuestran con claridad que los principales beneficiados por la autorización condicional son precisamente los grandes pagadores de la EMA. Del grupo de 21 multinacionales, hay tres que no cuentan con ninguna autorización de este tipo (Novo Nordisk, Accord y Teva). Las otras 18 acumulan en total 61 autorizaciones condicionales, lo que representa el 67% del total. La que más tiene es Janssen (9), seguida por Novartis (7), Roche (7), Pfizer (6) y AstraZeneca (5).
Por supuesto, esas 21 multinacionales son también las que más fármacos tienen en el mercado, así que se podría pensar que es lógico que acaparen dos de cada tres autorizaciones condicionales. No es así. Desde que empezó a funcionar la EMA en 1995, ha autorizado más de 1.700 fármacos, de los que pertenecen a las 21 multinacionales poco más de 800. Es decir, un 47%.
En tercer lugar, destaca el tiempo que tardan en ocasiones los laboratorios en presentar la evidencia de que su producto es eficaz y seguro. La autorización se otorga por un año y luego se puede ir renovando por el mismo periodo. En 2022, la EMA concedió el permiso estándar a dos fármacos que tenían autorización condicional desde 2012 (Caprelsa y Adcetris) y a otro que llevaba en el mercado desde 2013 (Bosulif). A día de hoy, aún están en el mercado cinco medicamentos que recibieron la autorización condicional en 2014 (Sirturo, Deltyba y Translarna) y 2016 (Ocaliva y una vacuna antigripal).
A preguntas de los periodistas, la portavoz de la EMA indicó que el tiempo medio que tarda un fármaco con autorización condicional en obtener la estándar “son tres años y ocho meses”. Pero los tiempos medios no siempre ofrecen la información más relevante. Investigadores del King’s College de Londres descubrieron que, entre 2013 y 2018, en la mitad de los casos las pruebas requeridas no se habían proporcionado más de siete años después de la autorización condicional. “Durante 30 años se nos ha dicho que los estudios posteriores a la comercialización llenarán los vacíos. Pero este no es el caso. No obtenemos estas pruebas”, lamenta Courtney Davis, sociólogo médico de la universidad británica.
La Agencia también defiende su política sobre las autorizaciones condicionales, que “sólo se recomiendan cuando el balance beneficio-riesgo del medicamento es positivo, el solicitante puede proporcionar datos completos tras la autorización, el fármaco satisface una necesidad médica no cubierta y el beneficio de la disponibilidad inmediata para los pacientes es mayor que los riesgos inherentes al hecho de que aún se requieren datos adicionales”.
Precisamente Ocaliva y Translarna, dos de los fármacos que llevan más tiempo en el mercado con una autorización condicional, están ahora bajo cuestión.
Ocaliva es un tratamiento para la cirrosis biliar primaria, una enfermedad hepática autoinmune. En octubre de 2023, la EMA inició “una revisión de beneficios y riesgos” del fármaco, “impulsada por los resultados finales de los dos estudios en pacientes” que había solicitado en 2016 como parte de los requisitos para conceder la autorización condicional. En otras palabras, los resultados finales tardaron siete años en llegar. Y no fueron precisamente positivos. Uno de los estudios, explica la portavoz de la EMA, “no demostró que Ocaliva fuera más eficaz que el placebo en cuanto al número de pacientes cuya enfermedad empeoró o que fallecieron. Además, los efectos secundarios, incluidos los graves, fueron más frecuentes en los pacientes tratados con Ocaliva”. La Agencia está revisando estos resultados, “junto con todos los demás datos disponibles”, antes de decidir si le retira la autorización a Advanz Pharma.
La Comisión Europea, a partir de las recomendaciones de la EMA, es quien toma formalmente las decisiones sobre los fármacos. A preguntas de Investigate Europe, un funcionario autorizado explicó que la Comisión “es consciente de las preocupaciones relacionadas con Ocaliva” y que el informe de la EMA se espera para este mes de junio. “Tan pronto como la Comisión reciba este dictamen, seguiremos la acción reguladora apropiada con respecto a la autorización de este producto”, concluyó.
Translarna, por su parte, es un medicamento para la distrofia muscular de Duchenne patentado por PTC Therapeutics. En el momento de la renovación anual en 2023, la EMA decidió que debía retirarse del mercado. Entre los efectos secundarios notificados figuran afecciones cardiacas graves. La Comisión Europea, que casi siempre sigue las recomendaciones de la EMA, no lo ha hecho en esta ocasión. En la respuesta que el funcionario ofreció a Investigate Europe se insinúa además la existencia de un conflicto de interés en este procedimiento: “La Comisión cree que la Agencia debería considerar las posibles implicaciones de la reciente sentencia del Tribunal de Justicia de la Unión Europea (asunto Hopveus), que afecta a la composición de los grupos científicos consultivos que participan en una evaluación y su conformidad con el principio de imparcialidad objetiva”. El funcionario se negó a ofrecer más información ante las preguntas de los periodistas.
El asunto Hopveus se refiere a un fármaco del mismo nombre que está indicado para tratar el síndrome de abstinencia alcohólica. La EMA negó su autorización y el fabricante, la compañía francesa D&A Pharma, recurrió a los tribunales europeos. El TJUE, en una sentencia conocida el pasado mes de marzo, le dio la razón y anuló la decisión negativa de la EMA al detectar un conflicto de interés en uno de los expertos consultados.
Los conflictos de interés de los expertos son, sin duda, otra carpeta abierta en la Agencia Europea de Medicamentos. Por Hopveus, por Translarna y por otros casos.