Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Agencias Reguladoras
Investigaciones
Solución para un sistema farmacéutico en crisis: recomendaciones para su reforma en Estados Unidos y Canadá (Healing an ailing pharmaceutical system: prescription for reform for United States and Canada)
Gaffney A, Lexchin J et al
BMJ 2018;361:k1039
Disponible en http://www.pnhp.org/Pharma
Traducido por Salud y Fármacos
Nuestros sistemas farmacéuticos no funcionan, y solo una reforma radical podría garantizar el acceso universal a medicamentos más seguros, más innovadores y más asequibles.
Mensajes clave
Introducción
Los medicamentos son una de las herramientas más poderosas de la medicina. Sin embargo, los sistemas farmacéuticos de EE UU y Canadá son totalmente disfuncionales. La forma en que la industria establece los precios- cobrando lo que el mercado aguante, especialmente en EE UU- rompe los presupuestos y hace que muchos pacientes no puedan acceder a medicamentos vitales [1-4]. A pesar de algunos avances importantes, la tasa de innovación real de la industria sigue siendo desproporcionada con nuestro elevado gasto en medicamentos; cada año se comercializan muchos medicamentos nuevos, pero pocos representan mejoras clínicas sustanciales [5-7]. Y los imperativos comerciales distorsionan los ensayos clínicos con medicamentos [8], las prioridades de investigación y la regulación de medicamentos [9,10].
Si bien muchos reconocen la necesidad de hacer cambios, las soluciones propuestas son diferentes [3,11-13] y no lograrían implementar los cambios fundamentales que estas deficiencias requieren. Médicos para un Programa Nacional de Salud organizó un grupo de trabajo compuesto por médicos, académicos y defensores de los consumidores de EE UU y Canadá (el Grupo de Trabajo para la Reforma de la Política Farmacéutica en EE UU / Canadá) para que elaborara una amplia propuesta de reforma para ambas naciones. Aunque las circunstancias políticas, incluyendo la influencia del lobby farmacéutico, hacen que la implementación completa de estas reformas sea improbable en este momento, cambios en los vientos políticos podrían generar un clima más favorable. Por lo tanto, el grupo de trabajo tuvo como objetivo elaborar una ambiciosa propuesta de reforma farmacéutica para establecer una agenda para el futuro, que incluye cobertura de seguros, precios, desarrollo de medicamentos, pruebas clínicas, aprobación regulatoria, monitoreo post-comercialización y promoción.
Si bien algunas de nuestras recomendaciones (recuadro 1) podrían implementarse dentro de la estructura que EE UU utiliza para financiar los servicios de salud, la implementación plena requeriría un sistema universal con financiador único. Canadá ya tiene un sistema con financiador único, pero requeriría reformas porque el sistema cubre todos los servicios hospitalarios y médicos, pero no los medicamentos que se consumen fuera del hospital [14,15].
Nuestra propuesta se basa en seis principios:
Acceso a medicamentos de venta con receta
En EE UU y Canadá se viola el derecho a medicamentos esenciales con frecuencia (figura 1). Los altos costos de bolsillo impiden que millones puedan surtir sus recetas [14,15,18] y llevan a muchos a la quiebra [19,20]. Se estima que en EE UU hay 28 millones de personas sin seguro médico [21], mientras que en Canadá 3,5 millones carecen de cobertura de medicamentos [14].
El costo compartido (copagos, deducibles y co-seguros) también impide el acceso en ambos lados de la frontera [15,22]. Limita la atención necesaria y la innecesaria en proporciones similares [23]; contribuye a que disminuya la adherencia [24,25]; y, para algunas patologías, agrava las disparidades raciales en la salud [26], eleva el gasto sanitario no relacionado con los medicamentos y empeora los resultados [24,26]. Cabe destacar que Gales, Irlanda del Norte y Escocia han logrado proporcionar cobertura universal de medicamentos sin compartir costos y utilizando otros mecanismos de control de costos para mantener el gasto en medicamentos muy por debajo de los niveles de EE UU o Canadá [27,28].
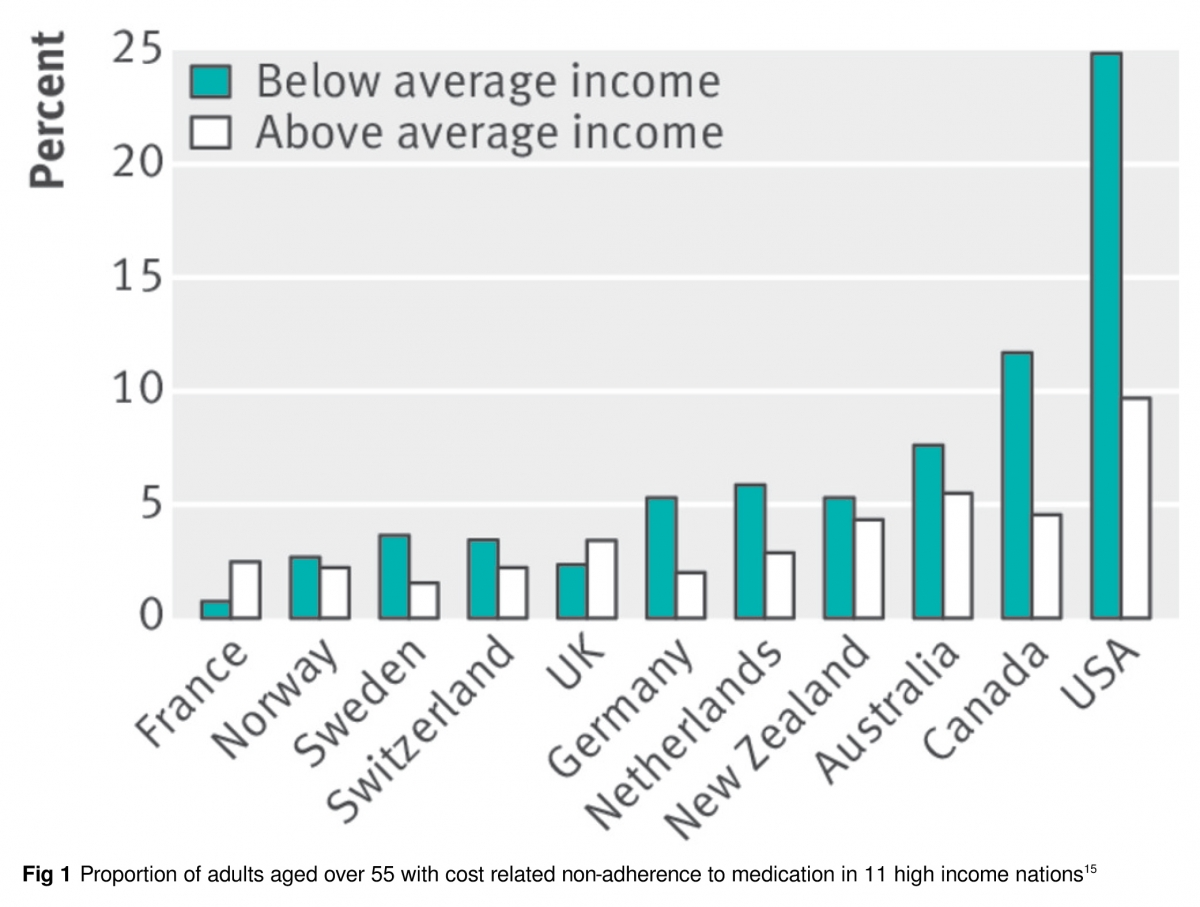
Para mejorar el acceso y la salud de la población, proponemos cobertura universal integra [29] (seguro completo sin compartir costos) de todos los medicamentos clínicamente necesarios, haciendo eco a la famosa invocación de Archie Cochrane: “todos los tratamientos efectivos deben ser gratuitos”[30]. Cada nación debería establecer un Formulario nacional de los medicamentos cubiertos, que debería incluir todos los medicamentos que han demostrado alargar o mejorar la calidad de vida, y cuando se disponga de varios productos equivalentes se debe incluir la opción más segura, más efectiva y menos costosa. Una oficina nacional de evaluación de tecnología podría proporcionar información de efectividad comparativa para orientar las decisiones sobre los productos que se incluyen en el formulario. Cuando sea clínicamente apropiado, por ejemplo, en caso de alergias u otras circunstancias únicas, los medicamentos fuera del formulario también deberían estar cubiertos.
Precios de los medicamentos
El gasto en medicamentos para consumo ambulatorio es más alto en EE UU (US$1026 anuales per cápita) y Canadá (US$713) que en otras naciones de la Organización para la Cooperación y el Desarrollo Económico (un promedio de US$515, y solo US$240 en Dinamarca) [27]. Estas diferencias se deben a los altos precios (especialmente en EE UU) y no a que se consuman más medicamentos. Por ejemplo, en 2014 una dosis diaria de 50 unidades de insulina glargina costaba US$186,38 al mes en EE UU (después de los descuentos correspondientes) frente a US$63,65 en el Reino Unido y US$46,60 en Francia [31].
A pesar de que hay quien afirma lo contrario, los costos de investigación y desarrollo no justifican estos altos precios [32]. Por ejemplo, el gasto total en investigación y desarrollo de 10 empresas que recientemente introdujeron nuevos medicamentos contra el cáncer ascendió a US$9.000 millones, mientras que esos medicamentos generaron US$67.000 millones en ingresos [33]. En EE UU, las compañías farmacéuticas, décadas después de haber recuperado los costos de desarrollo, siguen aumentando drásticamente los precios [1,34-36] y sus ganancias medias son consistentemente tres veces más altas que el promedio de las otras firmas incluidas en el Fortune 500: 23% v 7% respectivamente en 2016 [37].
Para reducir los precios de los medicamentos y garantizar que no se omite a ningún medicamento único se podrían dar varios pasos. La agencia reguladora de cada país seguiría aprobando los medicamentos sin importar el precio. Una vez aprobados, sin embargo, una agencia pública podría negociar los precios con los fabricantes, teniendo en cuenta (entre otras cosas) los datos comparativos de efectividad. La experiencia internacional y la de EE UU indica que tales negociaciones pueden reducir los precios [38], en EE UU probablemente lograría reducir los precios de los medicamentos de marca en alrededor del 50% [27,39-41].
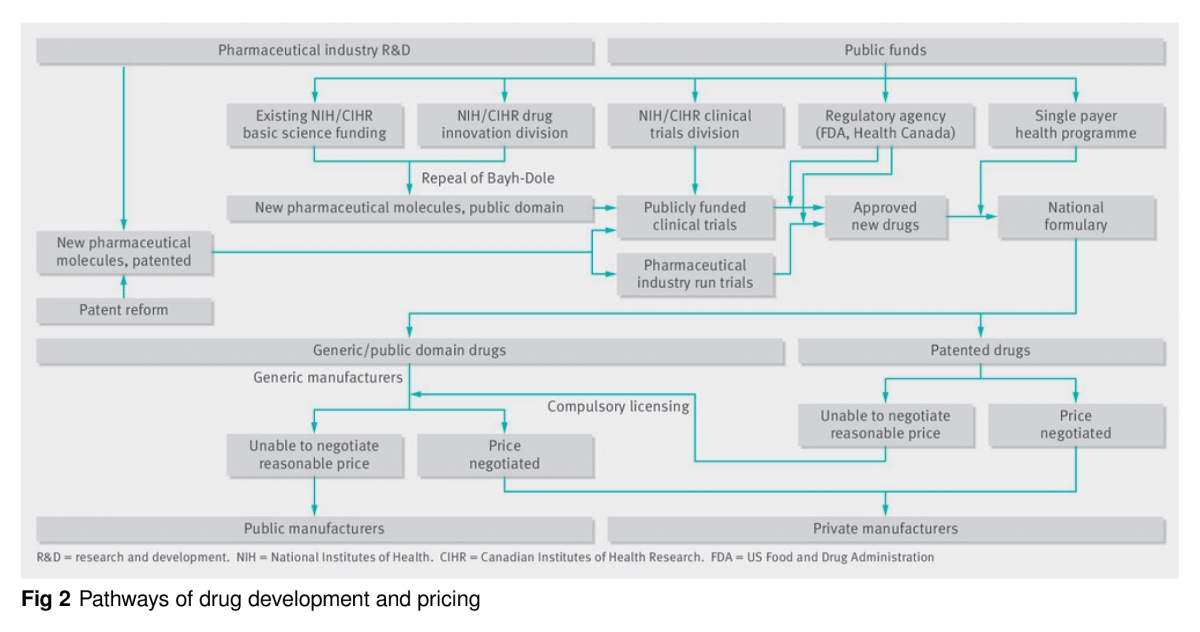
Si bien las negociaciones y el formulario nacional podrían reducir los precios de muchos medicamentos, cuando están protegidos por patentes y carecen de competencia, las empresas podrían seguir exigiendo precios irrazonables, obligando a los países a excluir el medicamento o revisar los presupuestos [42,43]. Por lo tanto, para asegurar precios razonables se requieren otras opciones (Figura 2). Por ejemplo, si fracasan las negociaciones sobre los precios de los medicamentos de marca, los gobiernos emitirían una licencia obligatoria para permitir la fabricación de genéricos, un mecanismo ya sancionado en virtud del derecho mercantil internacional [44], la ley de patentes de Estados Unidos [45] y la Ley Bayh-Dole [46]. De hecho, tanto las administraciones de Bush (EE UU) [44,47] como de Chretien (Canadá) [48] cuando en 2001 temían casos de bioterrorismo con ántrax, amenazaron con romper la patente de la ciprofloxacina, lo que hizo que Bayer bajara el precio.
Sin embargo, en algunas circunstancias, incluso las licencias obligatorias pueden no lograr precios razonables; el costo de algunos medicamentos genéricos se ha disparado desde que los únicos fabricantes de genéricos se hicieron con el mercado [1,35]. Por lo tanto, abogamos por generar capacidad para la producción pública de medicamentos cuando no se disponga de otras opciones a un precio razonable. Esta capacidad también serviría para aumentar la producción durante emergencias de salud pública o en periodos de escasez de medicamentos [49].
Finalmente, los medicamentos desarrollados con financiamiento público por entidades públicas no estarían protegidos por patentes y se podrían fabricar genéricos en todo el mundo a un costo muy reducido.
Desarrollo preclínico de medicamentos
La protección por patentes y la exclusividad en el mercado aumentan los precios de los medicamentos, pero típicamente se describen como fundamentales para fomentar la innovación. Esta representación es engañosa por dos razones.
En primer lugar, a pesar de haber logrado algunos avances importantes, el récord de innovación de la industria farmacéutica es irrisorio en comparación con sus enormes ingresos y ganancias [50]. La mayoría de los medicamentos nuevos tienen pocas ventajas sobre los tratamientos existentes y un precio mucho más elevado [2,6,7,51-55] mientras que las empresas a menudo amplían su exclusividad en el mercado a través de modificaciones triviales y patentes secundarias [56,57].
En segundo lugar, no está nada claro que las patentes sean el estímulo más importante para el avance terapéutico. A lo largo de la historia, a menudo ha sido la curiosidad y las recompensas intrínsecas al descubrimiento las que han impulsado los avances científicos, y no los incentivos financieros. Incluso hoy en día, la mayoría de las investigaciones básicas que subyacen a la innovación de medicamentos se llevan a cabo en instituciones públicas o sin fines de lucro, y las financian los Institutos Nacionales de Salud (NIH) y los Institutos Canadienses de Investigación en Salud (CIHR). En EE UU, antes de la Ley Bayh-Dole de 1980, los frutos de la investigación financiada con fondos públicos permanecían en el dominio público. Sin embargo, desde 1980, los investigadores financiados con fondos públicos pueden patentar sus descubrimientos y venderlos a firmas farmacéuticas [58], como ocurrió con el medicamento contra la hepatitis C, sofosbuvir. Aunque Bayh-Dole permite que el gobierno rompa las patentes de tales medicamentos, esta disposición nunca se ha usado [46].
Por lo tanto, proponemos la derogación de Bayh-Dole para mantener los medicamentos desarrollados con fondos públicos en el dominio público. Mientras tanto, hay que reformar el sistema de patentes para los medicamentos que desarrolla totalmente el sector privado, para que estimule la producción de medicamentos innovadores, no los productos parecidos los llamados me toos “yo también”.
En EE UU, los criterios para emitir patentes de medicamentos se han flexibilizado mucho más allá del requisito original de que un descubrimiento patentable tenía que ser útil, novedoso y no obvio [57,59]. Como otros han argumentado [3,60,61], las reformas al sistema de patentes podrían bajar los precios y promover la innovación. Las pequeñas variaciones o combinaciones de agentes existentes, isómeros de medicamentos [3] y ajustes en los dispositivos de administración de fármacos que no agregan una funcionalidad importante no deben ser patentables. Algunos países ya han impuesto restricciones similares [62].
Como las reformas que promovemos reducen los incentivos para que la industria desarrolle productos comercializables a partir de nuevos descubrimientos importantes, proponemos crear institutos para el desarrollo de medicamentos de venta con receta dentro de los NIH y CIHR. Los nuevos institutos tendrían dos divisiones: una para la innovación farmacológica y otra para los ensayos clínicos (figura 2). La división de innovación farmacológica se centraría en el desarrollo de agentes no patentables hasta cuando tuvieran que hacerse los ensayos clínicos. Esta “vía pública” junto con los fondos privados de investigación, desarrollaría nuevos productos farmacéuticos. Proponemos financiamiento público equivalente a aproximadamente la mitad de la inversión actual que hace el sector privado en la investigación preclínica. Todas las moléculas nuevas que desarrollase esta división permanecerían en el dominio público. Este enfoque es una forma de “desvinculación”, que otros han propuesto entre el desarrollo de los medicamentos y la fijación de sus precios [63,64].
Las divisiones de innovación de medicamentos podrían hacer algún tipo de desarrollo de fármacos, pero en general financiarían esfuerzos de investigadores académicos u otros investigadores no comerciales. Se daría prioridad a los medicamentos con el mayor valor clínico, centrándose en las enfermedades que se descuidan, comercialmente no rentables, que carecen de tratamientos efectivos o importantes para la salud pública. Las compañías de cualquier lugar del mundo podrían producir los nuevos medicamentos no patentados como genéricos, un gran avance para la salud mundial.
Pruebas clínicas
Los ensayos clínicos patrocinados por la industria a veces utilizan métodos poco sólidos e informan hallazgos incompletos, por lo que su interpretación, y algunas veces la veracidad de sus conclusiones sobre seguridad y eficacia, son cuestionables [8]. Por ejemplo, los ensayos han comparado los nuevos agentes con placebos en lugar de con las mejores terapias existentes, han utilizado dosis demasiado bajas del comparador, o han dependido de medidas subrogadas [65] que pueden no predecir los resultados clínicos. Algunos investigadores financiados comercialmente también han publicado selectivamente (y republicado) resultados positivos [8,66] u ocultado los hallazgos negativos [67], mientras que las empresas han interrumpido los ensayos prematuramente por razones puramente comerciales [68].
Mientras tanto, la propiedad corporativa de los datos de los ensayos clínicos puede enmascarar los problemas de seguridad e impedir futuras investigaciones [69]. Si bien el requisito de registro de los ensayos ha sido un importante avance, persisten los problemas de transparencia [70].
Por lo tanto, las agencias reguladoras de medicamentos deben elevar los estándares para presentar la evidencia. Los ensayos deben, siempre que sea posible, comparar los nuevos productos con las terapias existentes y utilizar un diseño de superioridad para desalentar la inversión en medicamentos innecesarios. Cuando los nuevos agentes sean imitaciones de los existentes, en general, deberán testarse en pacientes que no respondan (o toleren) los productos existentes. Y con pocas excepciones, los ensayos deben evaluar resultados clínicos sólidos (en lugar de medidas subrogadas) [71]. Los datos anonimizados de los pacientes y de todos los ensayos (incluyendo los más antiguos) deben ponerse a disposición del público [70] (independientemente de que el fármaco obtenga el permiso de comercialización) para facilitar la rendición de cuentas y la investigación.
Finalmente, debido a la preocupación por la objetividad de los ensayos financiados por la industria y la necesidad de probar terapias no patentadas y no rentables, las divisiones de ensayos clínicos de los nuevos institutos NIH e CIHR también deberían financiar y supervisar los ensayos (fig 2) [69,72]. Las divisiones seleccionarían las moléculas prometedoras desarrolladas por los laboratorios sin fines de lucro, los investigadores académicos y las compañías farmacéuticas para hacer los ensayos clínicos, que principalmente serían diseñados y realizados por investigadores no comerciales. También podrían financiar ensayos que evalúen nuevas indicaciones para productos no existentes o terapias no farmacológicas.
Los ensayos financiados con fondos públicos ofrecerían beneficios importantes: reducirían al mínimo los conflictos de intereses comerciales; redireccionarían la investigación de fármacos “yo también” hacia innovaciones reales, y facilitarían el desarrollo de tratamientos no rentables pero esenciales [69,72]. Aunque las empresas aún podrían financiar los ensayos de sus productos [72] porque los ensayos clínicos son costosos y estarían sujetos a un mayor escrutinio regulatorio (en base a la evidencia previa de empresas que manipulan los resultados), a largo plazo probablemente habría un predominio de ensayos financiados con fondos públicos.
Reforma al sistema de aprobación de medicamentos
Las agencias reguladoras canadienses y estadounidenses permiten con demasiada frecuencia que medicamentos peligrosos lleguen al mercado [73-76] y después de su comercialización los controlan inadecuadamente [77-79]. A partir de la década de 1990, la independencia de ambas agencias se ha visto erosionada por su dependencia de los pagos que hacen las compañías farmacéuticas. En EE UU, la recepción de fondos por parte de la FDA está explícitamente relacionada con el acortamiento de los tiempos de revisión de las solicitudes de comercialización [73,75].
Mientras tanto, una proporción cada vez mayor de medicamentos nuevos califican para participar en programas que reducen aún más los tiempos de revisión. En el 2014, el 69% de las solicitudes de comercialización de medicamentos presentadas a la FDA obtuvieron una “revisión acelerada” por diferentes designaciones o vías [80]. La cifra comparable para Canadá en el período 1997-2012 fue del 26% [81]. Estos programas, aunque fueron diseñados para acelerar la disponibilidad de agentes innovadores, han sido explotados para acelerar la comercialización de muchos medicamentos “yo también” o “me-too” [7,80]. Algunos métodos de revisión acelerada usan estándares de evidencia más débiles. La recientemente promulgada ley 21st Century Cures Act en EE UU crea aún más vías, y exige que la FDA evalúe el posible uso de “pruebas del mundo real”, es decir, no de ensayos clínicos, para aprobar nuevas indicaciones de medicamentos [82,83].
Estos cambios a lo que hay que presentar como evidencia puede aumentar el riesgo de que medicamentos inseguros se comercialicen [76,84]. Y la mayoría [73-75,85,86] de los estudios, pero no todos [84], sugieren que tiempos de revisión más cortos son perjudiciales.
Proponemos varias reformas al proceso de aprobación de medicamentos: en primer lugar, hay que acabar con el financiamiento de las agencias reguladoras de medicamentos por parte de la industria; los gobiernos deben financiar completamente los presupuestos de las agencias. En segundo lugar, la revisión acelerada debe reservarse para medicamentos que probablemente ofrezcan avances clínicos genuinos. Por ejemplo, los medicamentos “primeros en su clase” no deberían calificar automáticamente para la aprobación acelerada, ya que muchos no son superiores a los productos existentes [7]. En tercer lugar, el requisito de que los ensayos utilicen medidas de impacto clínico y comparadores activos solo debe suspenderse en circunstancias excepcionales. En cuarto lugar, si bien los expertos que reciben financiación comercial pueden ofrecer testimonios ante paneles asesores que evalúan medicamentos, no deben tener voto ni capacidad decisoria en esos paneles [87]. Finalmente, para que un medicamento sea elegible para obtener la exclusividad en el mercado, se debe exigir que demuestre superioridad sobre cualquier producto existente, ya sea en eficacia. seguridad, o conveniencia de dosificación o administración.
La vigilancia post-comercialización
Con frecuencia, las agencias reguladoras, a medida que han ido aprobando más medicamentos en base a criterios indirectos de evaluación y menos ensayos clínicos o más pequeños, han solicitado estudios de postcomercialización para confirmar los beneficios o excluir riesgos graves [77]. Pero, esta estrategia tiene serias deficiencias. Si bien los grandes estudios post comercialización son fundamentales para garantizar la seguridad (especialmente cuando los efectos secundarios son raros), no se deberían utilizar para debilitar los requisitos de seguridad para otorgar la aprobación inicial. Y aunque el uso de grandes bases de datos para la vigilancia farmacosanitaria (p. ej., El Sistema Centinela [Sentinel System] de la FDA) son prometedores, hasta ahora, sus resultados han sido modestos y no pueden sustituir a los ensayos clínicos [88].
Lamentablemente, la implementación de mecanismos para exigir que se hagan los estudios post comercialización que ha ordenado la FDA es laxa. La FDA no ha utilizado plenamente su autoridad para penalizar a las empresas que no completan dichos estudios [77,78] y Health Canada ha permitido que las empresas sigan comercializando medicamentos durante años sin completar los ensayos requeridos [79].
Proponemos varias reformas para mejorar la seguridad post comercialización. El financiamiento de la FDA y Health Canada para estos esfuerzos debería incrementarse y ser similar al presupuesto para la revisión de nuevas solicitudes de comercialización de medicamentos, y las oficinas de seguridad deberían tener la misma posición jerárquica en estas agencias que las oficinas encargadas de otorgar permisos de comercialización. Las oficinas de monitoreo de seguridad deben tener poder para ordenar, de forma independiente, la inclusión de advertencias de seguridad y la retirada del mercado de los medicamentos inseguros, y las agencias deben usar su autoridad legal de manera más agresiva para procesar a las compañías farmacéuticas que no completen los estudios postcomercialización requeridos a tiempo. Finalmente, la información sobre las demoras debe estar disponible públicamente.
Algunas de estas reformas podrían llevarse a cabo sin legislación: desde 2007, por ejemplo, la FDA ha tenido autoridad para penalizar a las empresas que no realizan oportunamente los estudios post comercialización. Sin embargo, no ha ejercido ese poder de manera significativa [78]. Legislación reciente permite que Health Canada imponga multas sustanciales a las compañías que no cumplen [89].
Promoción
La promoción de medicamentos, incluyendo las visitas de representantes de la industria a las oficinas de los médicos, consume anualmente miles de millones de dólares, más que el gasto total en educación de estudiantes de medicina en EE UU [90-92]; los gastos en ventas y mercadotecnia exceden los de investigación y desarrollo [13]. Además de desviar fondos que podrían ser mejor utilizados para desarrollar medicamentos que salvan vidas, tal promoción es a menudo engañosa o inexacta [93-95]. Esto es especialmente cierto para la publicidad directa al consumidor (DTC): que en EE UU está bastante extendida [96] y, afecta en forma atenuada a Canadá [97]. La publicidad que menciona la marca de un medicamento de venta con receta junto con su indicación está prohibida en todas las demás naciones desarrolladas, excepto en Nueva Zelanda.
El gasto promocional eclipsa los pequeños presupuestos de las divisiones de la FDA y Health Canada que regulan el marketing. La FDA está abrumada por el gran volumen de materiales para revisar [98,99] y Health Canada ha delegado una gran parte de la supervisión reguladora de la promoción a terceros [97].
Proponemos una gran ampliación de la revisión promocional. Las agencias reguladoras necesitan más recursos (y más predecibles) para llevar a cabo evaluaciones rigurosas de todos los materiales promocionales [99]. No deberían depender de fondos que impongan plazos a sus revisiones, porque puede fomentar revisiones indulgentes y, para evitar conflictos de intereses, el financiamiento debería provenir solo del gobierno [99].
Una mejor supervisión debe ir acompañada de sanciones más duras para los casos de promoción engañosa o fuera de etiqueta. En el pasado, incluso las multas enormes no han disuadido a las violaciones de la industria [100] porque, como señaló un experto, “cuando estás vendiendo US$1.000 millones por año o más de un medicamento, es muy tentador para la compañía simplemente ignorar la multa y mantener el exceso de velocidad”[101]. Por lo tanto, las autoridades deben estar facultadas para suspender el derecho de las empresas a promocionar sus productos o, en casos extremos, presentar denuncias penales contra los ejecutivos de las farmaceuticas.
Aunque también estamos a favor de prohibir la publicidad directa al consumidor y los visitadores médicos, las garantías constitucionales al “discurso comercial” pueden impedir tales prohibiciones en EE UU [102]. Sin embargo, otras herramientas son claramente constitucionales, como la eliminación los créditos fiscales para actividades promocionales; además, si hay tratamientos alternativos disponibles, los medicamentos promocionados de esta manera podrían excluirse de los formularios. Los visitadores médicos de la industria también podrían ser contrarrestados por “visitadores académicos”[103] sin ánimo de lucro para optimizar las prácticas prescriptivas de los médicos [104].
Finalmente, los fondos de la industria pueden sesgar la educación médica continua (CME) [105] y las guías clínicas [106]. Las autoridades que otorgan licencias profesionales no deberían contar los créditos de educación continúa financiada por la industria como parte de los créditos obligatorios. En cambio, la formación continuada podría ser coordinada e implementada por un organismo similar al NPS MedicineWise australiano (www.nps.org.au), mientras que el desarrollo de guías clínicas debería, como mínimo, seguir las recomendaciones delineadas por el Institute of Medicine [107].
Economía de un programa farmacéutico nacional
Aunque nuestra propuesta tendría grandes implicaciones económicas y presupuestarias, hacer un análisis detallado de esos efectos va más allá del alcance de este artículo. Otros han estimado que un plan farmacéutico nacional para Canadá podría ahorrar US$7.300 millones de los US$22.000 que se gastan actualmente en medicamentos de venta con receta, pero esta estimación no contemplaba las nuevas inversiones en investigación, desarrollo y las medidas regulatorias que proponemos [108]. En el caso de EE UU, creemos que si se implementaran los mecanismos descritos anteriormente, el ahorro en los precios de los medicamentos podría compensar completamente los costos adicionales de la cobertura universal de medicamentos, sin copagos, y las nuevas inversiones públicas que recomendamos.
Para conseguir el cambio
Jonas Salk, inventor de la vacuna contra la polio, evitó patentar, y declaró: “Usted ¿Podría patentar el sol?” Hoy, en cambio, la especulación reina con demasiada frecuencia, en detrimento de la salud de la población.
Nuestra propuesta exige una reorientación fundamental de la política de medicamentos: lograría que los medicamentos fueran más asequibles para los pacientes y la sociedad, promovería la innovación, fortalecería los esfuerzos para garantizar la seguridad y efectividad de los medicamentos y mejoraría la evidencia disponible para los prescriptores y el público. Como los medicamentos desarrollados a través de los mecanismos públicos que hemos propuesto permanecerían en el dominio público, se podrían producir en forma genérica en cualquier parte del mundo, beneficiando a muchas naciones.
Las reformas que defendemos enfrentan la oposición de grupos muy poderosos, especialmente de las compañías farmacéuticas, y solo las estadounidenses que están en el Fortune 500, en 2016, generaron ganancias totales de US$67.700 millones [37]. Sin embargo, la mayoría de los estadounidenses, tanto demócratas como republicanos, ahora están a favor de medidas gubernamentales para bajar los precios de los medicamentos [109] y el 91% de los canadienses apoyan un beneficio farmacéutico universal [110]. Estos son mandatos populares inequívocos para el cambio. La ruta entre el sentimiento y la emisión de políticas será, sin duda, ardua. Sin embargo, la historia está repleta de ejemplos de amplias reformas, a menudo favorecidas por cambios impredecibles en las circunstancias políticas, que superaron los intereses fuertemente establecidos. Apuntamos con esta propuesta a proporcionar un plan para la reforma que se anticipa y que podría generar cambios transformadores en los sistemas farmacéuticos de nuestros países.
|