Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Estudios de no inferioridad ¿Cuánta evidencia?
Elard Walter Quispe Mena1
1 Médico Reumatólogo del Hospital Central de la Fuerza Aérea del Perú y Clínica Good Hope – Miraflores
“Una cosa moderadamente buena… no es necesariamente buena” Thomas Paine
El objetivo habitual de los estudios clínicos controlados, aleatorizados es la demostración de la superioridad (o no igualdad) del tratamiento experimental frente a un tratamiento de referencia (o bien frente a un placebo). El objetivo es demostrar la eventual “superioridad” de un tratamiento respecto a otro, que es el patrón de oro para demostrar la eficacia de los medicamentos [1,2].
En ocasiones el interés del estudio es demostrar que dos tratamientos son equivalentes dentro de un determinado rango, o bien que el tratamiento experimental no es inferior. Es decir, el estudio se hace para demostrar que el nuevo tratamiento tiene efectos “similares” o “no inferiores” sobre un evento clínico relevante (mortalidad, hospitalizaciones, recidivas de tumores, etc.), y/o que puede proporcionar otras ventajas (facilidad de administración, menor costo, efectos adversos, etc.). Si se consigue demostrar equivalencia, se podrá autorizar la utilización del nuevo tratamiento como alternativa terapéutica para aquellos que no pueden recibir el medicamento original por presentar intolerancia o hipersensibilidad [3].
En resumen, se desea que la nueva estrategia, aún sin ser superior, conserve una “equivalencia clínica”. Desde el punto de vista estadístico, la demostración de “equivalencia” implica que la nueva intervención no es superior ni inferior. De tal manera, que cuando se diseña un estudio sobre la nueva intervención se estima que no superará al tratamiento previo, y lo único que pretende demostrar que no es inferior. Este diseño de estudio es el denominado de “no inferioridad”.
Concepto de equivalencia terapéutica [4]. Un fármaco es equivalente a otro cuando la diferencia de sus efectos terapéuticos se considera irrelevante desde el punto de vista clínico. En este tipo de estudios no se pretende nunca establecer la igualdad, sino la equivalencia, por lo que se asume que existe una diferencia entre los tratamientos, pero se considera que esta diferencia es pequeña y que no tiene importancia clínica.
Tipos de estudios para determinar equivalencia clínica. Existe el riesgo de que los estudios de no inferioridad se interpreten con los mismos criterios que los de superioridad, lo que lleva a confusiones y errores importantes en la interpretación de los mismos. Los estudios de no inferioridad tienen que diseñarse e interpretarse utilizando estrategias diferentes a las que se utilizan en los estudios de superioridad, y es necesario conocerlas para hacer una interpretación correcta de los resultados. Los estudios para determinar equivalencia clínica pueden agruparse en dos categorías: aquellos que permiten evidenciar equivalencia y aquellos que permiten estimar equivalencia. Sobre la base de la evidencia aportada por cada tipo de estudio, pueden clasificarse en cinco niveles de evidencia. Ver tabla 1
Tabla 1 Clasificación de la evidencia para determinar equivalencia [4]
|
Evidencia de Equivalencia |
– Nivel 1: Estudios directos entre dos fármacos, con diseño de equivalencia o de no inferioridad. |
|
Estimación de Equivalencia |
– Nivel 2: Estudios directos entre ambos fármacos, con diseño de superioridad – Nivel 3: Estudios diferentes frente a un tercer comparador común. |
Hipótesis estadística [4]. La hipótesis estadística en los ensayos de superioridad (llamada hipótesis nula, Ho) es que no hay diferencia entre los tratamientos; y la hipótesis alternativa (H1) es que sí existe diferencia. Como en estadística es imposible demostrar que algo es cierto siempre, lo que se intenta es demostrar que es falso, para lo cual sólo hay que mostrar que la hipótesis no se cumple en una circunstancia determinada. Así, lo que se logra, estadísticamente, no es demostrar la hipótesis alternativa, sino rechazar la hipótesis nula, lo que permite aceptar la H1: que en este caso, es que un tratamiento es superior al otro.
En los estudios de no inferioridad también partimos de una Ho, pero de forma inversa a los estudios de superioridad; es decir, la Ho es que los tratamientos son diferentes, y al rechazarla, nos llevará a abrazar la H1; que en este caso, será que los tratamientos son iguales; o mejor dicho, que la diferencia de eficacia entre ambos no se considera clínicamente relevante
|
En los estudios de no inferioridad; la hipótesis alternativa es que “la diferencia de eficacia entre ambos no se considera clínicamente relevante” |
Aspectos metodológicos de los estudios de no inferioridad [1,4,5,6,7,8,9]
Los estudios clínicos de no inferioridad se realizan cuando se considera que no es ético usar el placebo como control. Para establecer la no inferioridad se requieren tres atributos principales:
a. El tratamiento en estudio no debe ser inferior, desde el punto de vista terapéutico, al tratamiento del grupo control.
b. El tratamiento debería demostrar eficacia en un ensayo controlado con placebo, si éste se realizara.
c. El tratamiento ofrece ventajas adicionales de seguridad, tolerabilidad, costo y conveniencia.
El diseño de un estudio de no inferioridad debe tener en cuenta que se requiere definir adecuadamente a la población, utilizar un control activo probado, hacer el seguimiento adecuado de los sujetos que participan en el estudio, y debe tener un poder estadístico adecuado. Además hay que ejercer imparcialidad al hacer las comparaciones; es decir, que se realicen en términos equilibrados y justos, sin favorecer a uno por encima del otro (“honestidad en las comparaciones”) y hay que asegurarse de que el estudio cumple con los criterios de sensibilidad (sensibilidad del estudio).
Se dice que un estudio clínico presenta sensibilidad de estudio (assay sensivity) cuando es capaz de distinguir entre un tratamiento eficaz y un tratamiento de menor eficacia. Si en un estudio se detectan diferencias, ello es prueba suficiente de que el estudio tiene sensibilidad y, por tanto, se puede concluir a favor de uno de los tratamientos. Sin embargo, cuando no se detectan diferencias frente al control puede deberse a que la eficacia de los tratamientos es realmente similar o a la incapacidad del ensayo para detectar las diferencias.
Por esto, para evitar los riesgos en el diseño de este tipo de estudios se recomienda seguir los siguientes pasos:
1. Solidez de la evidencia histórica (control activo). Determinar si existe una evidencia histórica de que el tratamiento que se utilizará como control activo tiene un efecto estable y establecido con claridad. Sin esta determinación, no es posible demostrar eficacia a través de un estudio de no inferioridad, y no debe realizarse el estudio.
2. Similitud en el diseño. Se debe ser muy minucioso en la selección de la población, la presencia de terapias concomitantes y la elección de los puntos finales. Estos estudios deben ser muy similares al diseño de los estudios controlados contra placebo, a partir de los cuales se ha extraído la evidencia histórica de los efectos del fármaco que se utilizará como control activo.
3. Conducción y calidad del estudio. En los estudios de no inferioridad, la similitud de resultados entre tratamientos puede deberse a que ambos son igualmente útiles, pero también a que son igualmente inútiles. La tasa de atrición de los pacientes incluidos en la muestra debe ser baja, ya que si se pierde un número elevado de pacientes, los resultados pueden dar la falsa impresión de similitud. Si por ejemplo, el 80% de los pacientes suspendiera en el grupo que utiliza el nuevo tratamiento y/o en el grupo control activo, el estudio demostraría gran similitud en el impacto clínico. Al no existir un placebo real (si no un control activo), esto podría interpretarse erróneamente, afirmando que el nuevo tratamiento conserva el efecto histórico del control activo (es decir, que es igualmente útil), mientras que en realidad no ha podido ser evaluado por el escaso número de pacientes verdaderamente tratados.
En los ensayos de superioridad, la falta de cumplimiento del tratamiento (la denominada compliance al protocolo), conspira contra resultados significativos. En los estudios de no inferioridad, cuanto mayor sea la atrición de pacientes mayor será la similitud entre los tratamientos y la posibilidad de que los resultados sean significativos (es decir, los tratamientos son equivalentes o no inferiores). Por lo tanto, se debe garantizar una estricta adherencia, seguimiento y cumplimiento del protocolo y debe ser de alta calidad.
4. Selección adecuada de un margen de no inferioridad. En la misma definición del estudio de no inferioridad surge la principal dificultad conceptual: ¿qué criterio se adoptará para aceptar que la nueva intervención no es inferior a la previa? Se debe establecer un margen de no inferioridad aceptable, tomando en cuenta los datos históricos, las consideraciones estadísticas y la relevancia clínica.
4.1. Definir cuál es la magnitud de la máxima diferencia clínica que el investigador considera no se puede superar para considerar equivalentes los tratamientos, es decir, intervalo de valores de diferencia [en Reducción Relativa del Riesgo (RRR) o Reducción Absoluta del Riesgo (RAR) entre tratamientos], entre tratamientos, compatible con una diferencia sin importancia clínica. Es el llamado valor delta (∆).
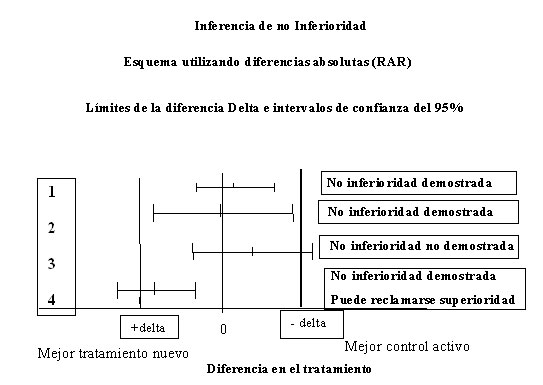
4.2. El valor delta. Cuando se estudia la equivalencia de dos o más tratamientos, uno de los pasos más importantes es definir qué se considera equivalente clínicamente. Debido a que la equivalencia absoluta es imposible de demostrar, lo que se hace es definir lo que se considera una diferencia irrelevante. El valor ∆ (delta), por lo tanto, no es un valor fijo, sino que hay que definirlo para cada caso, y es la máxima diferencia clínica que se puede tolerar para definir a dos tratamientos como equivalentes. Ver figura 1
Figura 1: Interpretación gráfica y conceptual de los análisis de no-inferioridad. Presentado por Tajer CD [3]
|
|
El parámetro graficado es la Reducción Absoluta de Riesgo (RAR): % del nuevo tratamiento – % del grupo del control activo (en cuatro estudios clínicos supuestos). En las abscisas se coloca el nivel de RAR = 0, es decir, ausencia de diferencia entre los grupos, y también el nivel ∆ (Delta), que se ha establecido como margen de no inferioridad.
Interpretación en ensayos de superioridad: los estudios 1, 2 y 3 muestran un incremento del riesgo con el tratamiento nuevo, en todos los casos no significativos. El cuarto muestra una reducción significativa con el tratamiento nuevo.
Interpretación en ensayos de no inferioridad: En el gráfico se establece un parámetro que no existe en los estudios de superioridad: el margen de diferencia que se considerará límite para la determinación de no inferioridad. En la medida en que el intervalo de confianza no cruce ese límite, podrá refutarse la hipótesis nula de inferioridad, y afirmar no inferioridad. Los estudios 1 y 2, si bien incrementan el riesgo, no alcanzan el margen preestablecido y por lo tanto queda demostrada no inferioridad. El intervalo de confianza del 3º cruza el margen, y por lo tanto no puede afirmarse no inferioridad. El estudio 4 también demuestra no inferioridad, pero dado que el IC no cruza siquiera el 0, puede plantearse que no solo no es inferior sino que, incluso resultó superior al control activo.
En este sentido, se han propuesto diferentes enfoques conceptuales:
a. Utilizar como valor delta el límite inferior de estudios previos (metanálisis incluidos) de la intervención clásica vs placebo. Esto puede expresarse en términos absolutos o relativos.
b. Utilizar un porcentaje que se considera clínicamente relevante: que la nueva intervención, por ejemplo, conserve como límite mínimo el 50 o el 80% del efecto de la clásica.
De acuerdo a la Conferencia Internacional de Armonización (ICH-E10 publicado en 1999), es fundamental que el valor delta se defina al comienzo del estudio y quede plasmado en forma clara en el protocolo, dado que se presta a manipulaciones fáciles, y correr el límite a posteriori puede provocar que incorrectamente se demuestre no inferioridad. Su valor es distinto para cada tipo de fármaco estudiado. En general, no existen criterios previamente establecidos de qué es importante clínicamente y que no; son las agencias de medicamentos, como la Food Drug Administration (FDA) y la Agencia Europea de Medicamentos (EMEA) las que lo determinan [10,11]. Por ejemplo, en los estudios clínicos de equivalencia y de no inferioridad de antirretrovirales se han aceptado valores delta del 10% al 12% de la variable del porcentaje de pacientes con carga viral indetectable a las 48 y 24 semanas.
|
La forma exacta de calcular el margen no se describe en el documento ICH-E10, y existe escasa experiencia publicada respecto a cómo efectuarlo |
4.3. Nunca el margen de no inferioridad puede superar el límite superior del intervalo de confianza del efecto de los ensayos previos. Siempre que se efectúa un estudio de tratamiento nuevo contra un control activo, subyace la idea de que al demostrar no inferioridad se pueda asumir que el tratamiento nuevo es también superior al placebo. Por ejemplo, si en un estudio originario el medicamento A redujo la mortalidad un RRR 15% (con un IC 95% de 5 a 25%) y riesgo relativo (RR) 0,85 (IC de 0.75 a 0,95) respecto del placebo, en ningún caso se aceptará como no inferior que en la comparación de C (control activo) vs el nuevo medicamento D no se conserve como mínimo ese 5%.
En los estudios de no inferioridad, el 1 será la incidencia de eventos del control activo, es decir, la medicación histórica. En nuestro ejemplo, para poder generar un margen de no inferioridad en términos de riesgo relativo, estableceremos que si el efecto histórico 0,85 fuera 1 y el extremo superior del IC el 0,95, expresados en términos relativos a ese 1 sería 0,95/0,85 = 1,12. Por lo tanto, si consideramos que para el próximo estudio el medicamento C (control activo) conserve su mismo efecto, el intervalo de confianza del riesgo relativo del medicamento D respecto de C no podrá ser superior a 1,12
|
“Suele ser razonable considerar que este margen (valor delta) sea entre un 5% y un 20% menor que la eficacia de referencia y ese margen debe estar plenamente justificado.” |
4.4. Algunos aspectos a tener en cuenta al valorar el margen de no inferioridad:
a. ¿cuál es el límite del intervalo de confianza, número absoluto o porcentaje respecto del efecto máximo?
b. Si el estudio resulta positivo, es decir, se afirma que el nuevo tratamiento no es inferior al control activo, puede deducirse en forma indirecta que aquel superaría al placebo en caso de que este estudio se efectuara. Esta afirmación es especulativa, para esto se requiere previamente responder a las siguientes interrogantes: ¿se ha definido adecuadamente el control activo? ¿el efecto del control activo se mantiene en forma constante? ¿el valor de no inferioridad es correcto?
5. Tamaño de la muestra. Mientras en los estudios de superioridad los resultados pueden tener intervalos de confianza (IC) grandes, siempre que sea estadísticamente significativo; en los estudios de no inferioridad, se debe exigir que los resultados y su IC (generalmente del 95%) estén dentro del rango establecido por delta. Dado que, el IC es menor cuanto mayor la muestra, resulta que el tamaño de la muestra en los estudios de no inferioridad es mayor que el requerido en los ensayos de superioridad.
En un estudio clásico de diferencias, el valor que se utiliza para calcular el tamaño de la muestra se basa en que el efecto sea lo suficientemente grande para que la diferencia entre los dos tratamientos resulte ser relevante. Pero en los estudios de no inferioridad, el valor delta debe ser un valor lo suficientemente pequeño para hacer irrelevante dicha diferencia. Como este valor exige que todos los resultados queden incluidos en este rango, los estudios de no inferioridad requieren mayor tamaño de la muestra para poder demostrar diferencias pequeñas. Las fórmulas que se utilizan para el cálculo del tamaño de la muestra son similares en los dos tipos de estudio.
El hecho de que un estudio de no inferioridad requiera mayor número de pacientes que uno de superioridad, implica que si durante un estudio de no inferioridad se demuestra superioridad de una de las ramas de tratamiento, éstas tendrán valor estadístico, ya que la muestra prevista será suficiente.
En resumen, durante la fase de diseño del estudio, se tratará de responder a algunas de las preguntas relacionadas con el tamaño de la muestra:
– ¿Cuántos individuos será necesario incluir en el estudio para mostrar que son equivalentes y garantizar la confirmación de la hipótesis alternativa?
– ¿Qué margen de equivalencia (o tamaño del efecto) podrán detectar la pruebas estadísticas si se incluyen en el estudio “n” sujetos?
– ¿Qué potencia tendrá la prueba estadística para detectar la hipótesis experimental si se incluyen en el estudio “n” sujetos?
Análisis de los resultados de los estudios de no inferioridad [1-7,12,13]
El análisis de los estudios de no inferioridad se basa fundamentalmente en los puntos anteriormente revisados y tiene aspectos diferentes a los estudios de superioridad. Se recomienda evaluar los siguientes aspectos:
1. Intervalo de confianza. Los resultados de las variables vienen dados por un valor medio y unos valores de intervalo de confianza (IC), que indican el rango dentro del cual está el verdadero resultado con una probabilidad determinada de certeza (generalmente 95%).
Para poder establecer equivalencia terapéutica, todos los valores que engloba el IC del 95% de la diferencia deben estar dentro de los límites del valor delta. Si alguno de los extremos del IC supera los valores de delta NO podrá establecerse la equivalencia o no inferioridad entre los tratamientos. Ver figura 1.
2. El valor de p. En los ensayos de superioridad, al rechazar la hipótesis nula (Ho) con un valor de p< 0.05, significa rechazarlo con un error del 5% (error alfa). Si p> 0,05 no se puede rechazar la Ho, pero tampoco afirmar, y nos quedamos en una zona de incertidumbre. Así, el valor de p es definitivo en la interpretación de los estudios de superioridad. Sin embargo, en los estudios de equivalencia, el valor de p carece de importancia, pues si no se muestra una diferencia (p> 0,05) no indica que exista equivalencia.
Como lo que se observa en los estudios de no inferioridad es que los resultados queden dentro de un margen previamente establecido, no importa que sean significativos o no. Puede haber resultados estadísticamente significativos con p< 0,05, pero que queden dentro del margen establecido como clínicamente equivalente y, por lo tanto, se consideran equivalentes
|
En los estudios de no inferioridad el valor de p no tiene importancia y lo que se debe valorar son los IC de los resultados obtenidos sean o no significativos. |
3. Análisis de las variables. El análisis de resultados de un estudio de superioridad se recomienda que se realice por “intención de tratar”, así se homogenizan los grupos y se dificulta encontrar diferencias, con lo que se garantiza la posición más conservadora, que permitirá valorar los resultados en el caso más desfavorable.
Sin embargo en los ensayos clínicos de no inferioridad, es necesario también realizar un análisis “per protocol” o de datos observados; es decir, de los pacientes que han sido asignados a un grupo, que cumplen los criterios de inclusión y exclusión, que han recibido el tratamiento completo y a los que se ha dado seguimiento hasta el final del estudio.
Con este enfoque aumentarán las diferencias entre tratamientos, lo que dificulta poder concluir que dos tratamientos son equivalentes, manteniendo la posición más cauta en la interpretación de los resultados. Si los resultados de los dos tipos de análisis no coinciden, es necesario investigar y analizar los subgrupos de pacientes que se han desviado del protocolo e investigar las causas, antes de llegar a conclusiones determinantes de equivalencia o no inferioridad.
|
Es importante que se produzca el mínimo número de pérdidas o abandonos de pacientes y las condiciones del seguimiento deben ser muy estrictas |
4. Sensibilidad del estudio. Como se señaló anteriormente en los estudios de no inferioridad, que dos medicamentos resulten ser equivalentes no implica que sean eficaces, ya que es igualmente compatible con la idea que ninguno es eficaz.
La sensibilidad se valorará por la evidencia histórica y el correcto diseño y seguimiento del estudio, por lo que es necesario que en los estudios que no sean de superioridad se realicen los siguientes pasos:
a. Determinar la evidencia histórica de la eficacia del medicamento con que se compara.
b. Que el control con que se compara haya probado ser superior a placebo.
c. Que la eficacia del control se mantenga en las condiciones del nuevo estudio.
d. La evaluación del valor delta.
Evaluar la validez interna del estudio.
|
La sensibilidad del estudio depende de: la evidencia histórica de eficacia el correcto diseño y seguimiento del estudio |
5. Aspectos éticos. Como en cualquier estudio clínico, los de no inferioridad, deben respetar escrupulosamente los principios éticos de autonomía, beneficencia, no maleficencia y justicia.
En los estudios de equivalencia o de no inferioridad, resulta difícil explicar a los candidatos a participar en el estudio que no existen ventajas esperables por su participan en el mismo y que no se espera obtener nada mejor por ensayar un fármaco nuevo, pero esto debe estar señalado en el consentimiento informado. El Comité de Ética es el responsable que esto se cumpla.
Los autores deben declarar sus potenciales conflictos de intereses (en especial mencionar los aportes para investigación, si son o han sido consultores de laboratorios farmacéuticos, y si son o no empleados del laboratorio patrocinador)
¿Cuál es la calidad metodológica de los estudios de equivalencia y de no inferioridad? [14,15]
La medicina basada en la evidencia (MBE) ha hecho que se cambie la forma de comunicar los resultados de los estudios clínicos, e incluso la forma de diseñarlos. Así, paralelamente a la aceptación universal de la MBE, se ha ido construyendo la ciencia del maquillaje de los estudios clínicos, cuyo objetivo es la introducción de nuevos tratamientos –sobre todo fármacos- en la práctica clínica. Una de las muchas consecuencias es que avanzamos hacia una sociedad cada vez más medicalizada, donde casi nadie va a librarse de recibir tratamiento, no ya por una enfermedad que afecte su calidad de vida, sino por tener uno o varios factores de riesgo.
La MBE propone tres preguntas claves para evaluar un estudio clínico y requiere una respuesta afirmativa a todas ellas para aplicar en la práctica sus conclusiones, estas son: ¿son fiables los resultados (validez interna)?, ¿son relevantes (magnitud del efecto)?, ¿son aplicables a mi paciente o contexto (validez externa)?
La validez interna se basa en la calidad metodológica del estudio, para el caso de los estudios de no inferioridad serían:
a. Una buena definición de la población a estudiar
b. Un control activo probado en ensayos clínicos y/o metanálisis(clínicamente relevante)
c. El valor ∆ (Delta) o margen de no inferioridad (su intervalo de confianza), con relevancia estadística pero fundamentalmente clínica, definida al comienzo del estudio.
d. El tamaño de la muestra cuidadosamente calculado (evitando el sesgo).
e. El seguimiento de los tratamientos, sin desviación del protocolo y optimizando la adherencia (evitar la atrición de pacientes).
f. La evaluación de las variables: por protocolo (de aquellos que han recibido el tratamiento completo y a los que se ha dado seguimiento hasta el final del estudio) y por intención de tratar. Siempre, revisando los resultados relativos vs los absolutos.
Presentaré algunos estudios que analizan estos aspectos:
– Greene WL y col [16]. En una revisión 88 estudios clínicos, publicados entre 1992 y 1996, que decían demostrar equivalencia: en el 51% se encontró equivalencia, pero sólo en el 23% fueron dentro del intervalo de confianza preestablecido, y sólo en el 22% se adoptó apropiadamente un margen de equivalencia predefinido.
– Krysan y Kemper [17], al revisar 25 estudios clínicos de equivalencia, publicados entre 1980 y 2000 sobre el tratamiento de meningitis bacteriana en niños, encontró que sólo en 2 estudios (8%) se definió adecuadamente el margen de no inferioridad.
– Costa LJ y col. [18] revisaron 188 estudios clínicos con resultados negativos usando un análisis de no inferioridad o equivalencia, sólo en 3 estudios se definió previamente el margen de no inferioridad.
– Piaggio y Pinol [19] evaluaron 20 estudios sobre salud reproductiva, sólo en 4 se estableció claramente el margen de no inferioridad.
– McAlister y Sackett [20] estudiaron 4 estudios clínicos de no inferioridad de los tratamientos de la hipertensión arterial, sobre el control activo. En dos se realizó tanto el análisis por intención de tratar como por protocolo, sólo en uno se preespecificó el margen de no inferioridad.
– Kaul y Diamond [1] analizó ocho ensayos clínicos de no inferioridad y valoró a la población, el control activo, la observancia del tratamiento y el poder estadístico. Encontró que sólo cuatro cumplían estos requisitos.
Estas evidencias ilustran lo complejo que puede ser la lectura crítica de los estudios de no inferioridad y concluir que efectivamente sí existe equivalencia o no inferioridad entre dos alternativas terapéuticas.
Conflicto de intereses: Participación en le curso Medicina Basada en Evidencias, programado por el laboratorio Aventis Pharma Perú. Participación en Congresos y Cursos Nacionales e Internacionales, financiado por los laboratios Merck Sharp & Dhome, Pfizer, Ely Lilly, Schering-Plough, Boehringer Ingelheim. Miembro de la red DURG-LA (Drug Utilization Research Group – Latin America) y AIS (Acción Internacional para la Salud).
Agradecimientos: Al profesor Joan-Ramón Laporte Roselló y Dr. Albert Figueras Suñé, por sus valiosos aportes y comentarios al manuscrito, ambos de la Fundación Institut Catalá de Farmacología, Hospital Vall Hebron, Barcelona-España. A la Dra. Fresia Castro Pavéz, Médica Reumatóloga de la Sección de Reumatología del Hospital Central de la Fuerza Aérea del Perú, por su acuciosa revisión.
Para contactarse: Correo electrónico, ewquispe@hotmail.com; ewquispe@yahoo.com
Referencias
1. Klaus S, Diamond GA: Good enough: a primer on the análisis and interpretation of noninferiority trials. Annals of Internal Medicine 2006;145:62-69
2. D´Agostino Rb, Massaro JM, Sullivan LM. Non-inferiority trials: design concepts and issues – the encounters of academic consultants in statics. Statist Med 2003;22:169-186
3. Tajer CD. Estudios de no inferioridad (Equivalentes terapéuticos: Concepto y niveles de evidencia). www.gedic.com.ar/publicaciones/Equivalencia_y_no_inferioridad.doc. Acceso verificado hasta el 4 de enero de 2008.
4. Delgado O, Puigventós F.: Diseño y evaluación de los ensayos de equivalencia. El Comprimido 2006;6:10-17. www.elcomprimido.com/articulos%20PDF/El%20Comprimido_n_6.pdf . Acceso verificado hasta el 04 de enero de 2008.
5. Pinteño M, Martínez-López I, Delgado O. Equivalentes terapéuticos: concepto y niveles de evidencia. El Comprimido 2006;6:14-18. www.elcomprimido.com/articulos%20PDF/El%20Comprimido_n_6.pdf. Acceso verificado hasta el 4 de enero de 2008.
6. Piaggio G, Elbourne D, Altaman D, Pocock S, Evans S for the CONSORT Group. Reporting of noninferiority and equivalence randomized trials. An extension of the CONSORT statement. JAMA 2006;295(10):1152-1160.
7. Henanff A, Giraudeau B, Baron G, Ravaud P. Quality of reporting of noninferiority and equivalence ramdomized trials. JAMA 2006;295(10):1147-1151.
8. Gotzsche P. Lessons from and cautation about noninferiority and equivalence randomized trials. JAMA 2006;295(10):1172-1174.
9. Sust M, Videla S. Control de sesgos en los diseños de equivalencia y no-inferioridad. Investigación Clínica y Bioética 2003, N° 15.
10. The European Agency for the evaluation of medicinal products evaluation of medicines for human use. Points to consider on switching between superiority and non-onferiority. London, 27 July 2000.
11. The European Agency for the Evaluation of Medicinal Products Evaluation of Medicines for Human Use. Points to consider on switching between superiority and non-onferiority. Londres, 27 de julio de 2005.
12. Appel LJ. A Primer on the design, conduct, and interpretation of clinical trials. Clin J AM Soc Nephrol 2006;1:1360-1367.
13. Pater C. Equivalence and noninferiority trials – are they viable alternatives for registration of new drugs? Current Controlled Trials in Cardiovascular Medicine 2004;5:8.
14. Márquez S, Meneu R. La medicalización de la vida y sus protagonistas. Gestión Clínica y Sanitaria 2003;5:47-53.
15. Peiró S. La construcción y comunicación del conocimiento en la era de la medicina basada en evidencia. Implicaciones para la lectura crítica de ensayos clínicos. Gestión Clínica y Sanitaria 2006;8:87-91.
16. Greene WL, Concato J, Feinstein AR. Claims of equivalence in medical research: are they supported by the evidence? Ann Intern Med 2000;132:715-722.
17. Krysan DJ, Kemper Ar. Claims of equivalence in randomized controlled trials of the treatment of bacterial meningitis in children. Pediatr Infect Dis J 2002;21:753-758.
18. Costa LJ, Xavier ACG, del Giglio A. Negative results in cancer clinical trials – equivalence or poor accrual? Control Clin Trilal 2004;25:525-33
19. Piaggio G, Pinol AP. Use of the equivalence approach in reproductive health clinical trials. Stat Med 2001;20:3571-577
20. Mac Alister FA, Sackett DL, Active – control equivalence trials and antihypertensive agents. Am J Med 2001;111:553-558
(regresa a regulación_y_políticas)