Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
Agencias Reguladoras
América Latina
Argentina. Respiradores y dispositivos quirúrgicos ingresaron al país pese a haber sido retirados en EEUU por riesgo de muerte
Mariel Fitz Patrick
Infoabe, 25 de noviembre de 2018
http://www.infobae.com/sociedad/2018/11/25/respiradores-y-dispositivos-quirurgicos-ingresaron-al-pais-pese-a-haber-sido-retirados-en-eeuu-por-riesgo-de-muerte/
Fueron importados en Argentina pese a la alerta sanitaria internacional. La información era considerada hasta ahora “confidencial” por la ANMAT
Al menos una docena de productos médicos importados en el mercado argentino debieron ser retirados o corregidos por sus fabricantes extranjeros, a partir de que se detectaran fallas con potencial peligro de muerte para los pacientes. Entre ellos respiradores y dispositivos médicos para el corazón. Varios ingresaron al país incluso después que saltaran las alertas sanitarias en EE UU.
Esta información que hoy revela por primera vez en Argentina la investigación The Implant Files, del Consorcio Internacional de Periodistas de Investigación (ICIJ), hasta ahora no se conocía públicamente. Esto era así porque, según la Administración Nacional de Alimentos, Medicamentos y Tecnología Médica (ANMAT), los procedimientos llevados a cabo en cada caso son “confidenciales”.
La lista incluye desde dispositivos cardíacos y vasculares, hasta sistemas para asistir a los médicos durante las cirugías cardíacas o de cráneo, respiradores y equipos de diagnóstico por imágenes.
El equipo argentino de ICIJ – que integran Infobae, La Nación y Perfil– reconstruyó esta información a partir de cruzar la base de datos públicos de la FDA que fueron sistematizados por ICIJ, con los registros de importaciones de la Aduana argentina, la información aportada por la ANMAT y las respuestas de las empresas importadoras a las consultas de los periodistas argentinos.
En Argentina no hay cifras oficiales sobre la cantidad de prótesis y dispositivos médicos que debieron ser retirados o corregidos en el mercado, hospitales e instituciones. En el área de Tecnovigilancia de la ANMAT, que se ocupa de los efectos adversos en los Productos Médicos (PM) autorizados en el país, explicaron que “están trabajando en estadísticas sobre los efectos adversos reportados”.
La FDA, la autoridad sanitaria de referencia en el mundo, publica diariamente lo que se denomina recalls o advertencias sobre distintos implantes y dispositivos médicos. Lo hace a partir de la información que brinda un fabricante ante la sospecha o certeza de que un producto puede tener una falla o provocar un riesgo a la salud. Según su gravedad, los califica de I a IV, siendo los de Clase I, los de mayor riesgo potencial, incluida la muerte.
Alertas con riesgo de muerte
El equipo argentino de ICIJ se concentró en identificar qué productos médicos con recall Clase 1 de la FDA se comercializaron en Argentina una vez emitida la alerta global. Para eso, revisó las autorizaciones de la ANMAT y buscó averiguar qué se hizo respecto de ellos una vez ingresados al mercado. Esa información – a diferencia de lo que sucede con la página web de la FDA- no está accesible en el sitio del organismo sanitario argentino.
Tampoco fue posible encontrar las disposiciones de la ANMAT publicadas en el Boletín Oficial con el aviso de retiro o la acción correctiva dispuesta sobre un implante o dispositivo, como sí sucede habitualmente con un medicamento, alimento o artículo cosmético considerados nocivos para la población.
Desde el organismo, sostuvieron que solo emiten una disposición pública “cuando hay una amenaza seria a la salud pública”. Explicaron que no lo hacen “si son casos muy puntuales de efectos adversos, para no alarmar a la población”.
También aclararon que un recall, no siempre significa el retiro del mercado del producto, sino que en ocasiones puede implicar que el dispositivo médico -si es un equipo para cirugía o de diagnóstico, por ejemplo- necesita ser revisado o arreglado. Cuando se trata de un implante ya colocado en el cuerpo -como una prótesis de cadera o malla quirúrgica, por ejemplo – no siempre puede ser extraído fácilmente de los pacientes. En ese caso, la fabricante delega en los médicos que se pongan en contacto con sus pacientes para decidir los pasos a seguir, en función del costo-beneficio de una nueva intervención.
Pese a la reticencia inicial de la ANMAT en dar precisiones sobre qué pasó en cada caso, finalmente ante la insistencia periodística, admitió que en la mayoría de los casos consultados las importadoras tuvieron que realizar alguna “acción de campo” correctiva o de retiro del producto ante la alerta sanitaria. Esta información fue corroborada por las compañías involucradas que accedieron a responder las consultas del equipo del ICIJ.
Entre los casos detectados figuran dispositivos cardiovasculares que, tras la alerta, tuvieron que ser parcialmente retirados y destruidos; bombas para el corazón que resultaron ser riesgosas y equipos de diagnóstico o cirugía que debieron ser revisados y reparados cuando ya estaban en funcionamiento.
El gigante Medtronic
Medtronic, una empresa estadounidense líder en dispositivos cardiovasculares, es una de que más retiros del mercado debió hacer en este país y en el mundo. Luego de que, en los Estados Unidos, Medtronic informara que varios de sus productos tenían fallas que podían llegar a ocasionar la muerte de sus pacientes, esos mismos dispositivos destinados a personas con problemas cardíacos ingresaron y se distribuyeron en la Argentina.
Uno de ellos es el dispositivo de recuperación Alligator Retriever, una pinza de platino diseñada para extraer cuerpos extraños de los vasos sensibles (venas, arterias y capilares). Se utiliza en casos de aneurisma y es una herramienta quirúrgica desechable de un solo uso. El problema detectado por la empresa fue la deslaminación y desprendimiento del material de recubrimiento en el flujo sanguíneo del paciente, con riesgo de muerte.
La FDA emitió una primera alerta Clase 1 el 16 de abril de 2014 sobre 43 lotes, y un segundo aviso el 9 de noviembre de 2016 sobre otros 8 modelos del Alligator. Sin embargo, fue autorizado por la ANMAT para comercializarse en el país en abril de 2015.
Según los registros de Aduana, se importó el producto 18 días después de la segunda alerta de las autoridades sanitarias de los Estados Unidos, y mientras estaba vigente. “Existió una acción de campo”, se limitó a responder la ANMAT sobre el dispositivo Alligator.
Ante la consulta del equipo argentino de ICIJ, Medtronic brindó más datos: “Todas las unidades que ingresaron a Argentina afectadas por el aviso de la FDA fueron importadas y vendidas por Biosud SA. El 7 de octubre de 2016, Biosud notificó a la Anmat sobre este recall y se comunicó con las instituciones de atención médica que habían adquirido el producto para solicitarles que lo devolvieran. Los dispositivos fueron destruidos. El distribuidor proporcionó evidencia de la destrucción el día 14 de febrero de 2017 a Medtronic y a la ANMAT”.
Desde Biosud, sin embargo, señalaron: “Nosotros ya no teníamos la representación de Medtronic cuando salió el recall. La acción le correspondía a Medtronic y colaboramos brindando información de los clientes”.
El Alligator no fue el único dispositivo que Medtronic debió retirar del mercado argentino. El 15 de noviembre de 2013 la FDA emitió una alerta Clase 1 sobre varios modelos de alambres guías orientables de Medtronic Vascular que se utilizan para colocar un catéter de ablación y permitir el mejor acceso al corazón del paciente durante la cirugía. Fue después que el gigante de la industria médica hubiera advertido que el recubrimiento de teflón se “descaramaba”, como las espinas de un pescado. El desperfecto podía causar la muerte de los pacientes.
La ANMAT había autorizado un año antes a Medtronic Latin America INC la importación de varios modelos de esos alambres guía que debieron ser retirados del mercado en EE UU: Zinger, Cougar Nitinol y Attain Hybrid.
Desde la autorización oficial y hasta junio de 2018 ingresaron a la Argentina más de 2000 unidades de distintos modelos, a pesar de la vigencia de la alerta sanitaria de EE UU. La Anmat sostuvo ante la consulta periodística que el importador “retiró el producto del mercado” argentino.
Desde Medtronic aseguraron que solo “40 unidades de este producto importadas a la Argentina resultaron afectados por este recall” y, según pudo saber el equipo argentino de ICIJ, 34 unidades fueron devueltas a la empresa el 21 de noviembre de 2013 para ser destruidas. Otras 6 unidades ya habían sido utilizadas para el momento en el que llegó la alerta de la FDA. “No se recibieron observaciones o reclamos por parte de las instituciones de salud que las utilizaron”, dijo la empresa.
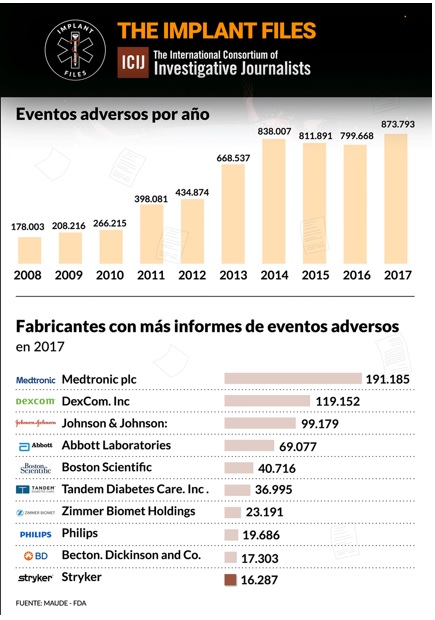
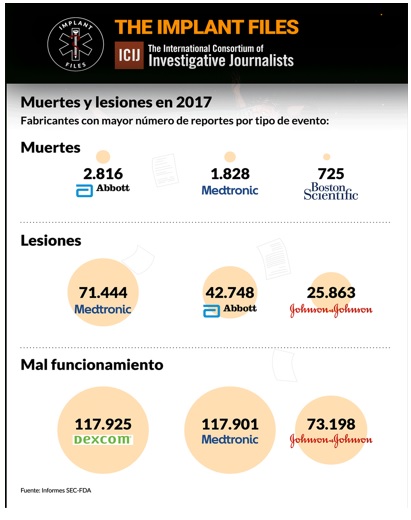
Un análisis de ICIJ de los informes de eventos adversos presentados ante las autoridades regulatorias de EE UU revela que Medtronic es el fabricante de dispositivos médicos con más efectos adversos reportados el año pasado: 191.185. De esta cifra, 1.828 se vincularon oficialmente a muertes, 71.444 a lesiones, y 117.901 a mal funcionamiento de los dispositivos.
Consultado por el Consorcio para The Implant Files, el director ejecutivo de Medtronic, Omar Ishrak, no quiso hacer comentarios. En una declaración escrita, el vocero Rob Clark dijo que la compañía considera la seguridad del paciente como su principal prioridad y respalda los “estándares más altos de práctica ética”.
Equipos de diagnóstico General Electric
Otra de las compañías estadounidenses con alertas por fallas en sus dispositivos médicos es General Electric. A través de su división GE Healthcare, fabrica grandes equipamientos de diagnóstico de uso externo que también pueden incidir en la salud de los pacientes. Cuatro modelos de equipos de esta marca que fueron importados a la Argentina tuvieron advertencia de riesgo de la FDA por fallas técnicas en alguno de sus componentes.
El equipo de diagnóstico Infinia Hawkeye 4 Nuclear Medicine System recibió una alerta sanitaria de la FDA el 24 de julio de 2013 porque una unidad de este modelo causó la muerte de un paciente en EE UU. “GE notó un incidente en una instalación de un centro médico de los Estados Unidos en el que un paciente falleció debido a las lesiones sufridas mientras se escaneaba en un Infinia”, señaló la alerta sanitaria.
La ANMAT había autorizado la importación de este equipo dos años antes, en junio de 2011, y llamativamente, volvió a autorizarlo en abril de 2014, cuando la alerta seguía vigente en EE UU. Culminó dos meses después.
A la Argentina, según registros de Aduana, ingresaron dos dispositivos similares en febrero y abril de 2013, y otro en octubre de 2014. “Hubo un servicio técnico para verificar si había un problema con los equipos a modo de prevención”, señalaron en ANMAT.
Como consecuencia del incidente con el equipo Infinia y por prevención, la FDA también publicó para la misma fecha una alerta para otro equipo de la misma marca, el Discovery NM 630. Esa alerta sanitaria en EE UU estuvo vigente entre el 24 de julio de 2013 y el 16 de junio de 2014.
Un equipo de este modelo fue importado a la Argentina el mes anterior a que se emitiera el aviso de la FDA y otra unidad, en diciembre de 2014, una vez superada la alerta internacional.
Lo mismo hizo General Electric con otro equipo, un sistema de tomografía computarizada Brivo NM615, diseñado para procedimientos de imágenes de medicina nuclear general con fines diagnósticos.
A partir del incidente con el Infinia, la FDA emitió otra alerta para este modelo en julio de 2013, vigente por un año. La ANMAT autorizó la importación en septiembre de 2012 a la importadora GE Healthcare Argentina, permiso que fue renovado en agosto de 2016. El equipo fue importado al país en octubre de 2015.
Ante la consulta del equipo argentina de ICIJ, General Electric respondió que “los retiros fueron provocados por un incidente aislado en EE UU. Como resultado, GE Healthcare realizó de inmediato una investigación exhaustiva de los dispositivos relevantes en todo el mundo, incluidas todas las unidades en Argentina. Confirmamos que no hubo un riesgo similar para los pacientes y, por lo tanto, no es necesario modificar ninguno de los dispositivos en Argentina”.
Tiempo después del episodio con Infinia, la FDA emitió otra alerta en febrero de 2015 por un potencial problema eléctrico en numerosos modelos de sistemas de resonancia magnética de GE Healthcare, entre otros, el Discovery MR450, el Discovery MR 750, y varios modelos Signa y Optima.
Según los registros de Aduana, al menos una unidad del modelo MR 750 fue importado en el país por GE Healthcare Argentina en septiembre de 2015. También ese año entraron cinco modelos Signa y distintas partes de los modelos Optima.
Al ser consultados por estos modelos reportados por la FDA, en General Electric respondieron que “la falla fue el resultado de un error del usuario y no un defecto del producto. Para garantizar que este error humano no volviera a suceder, GE Healthcare envió ingenieros de servicio para inspeccionar sus dispositivos en todos los sitios del mundo donde estos se encontraban instalados”.
Dispositivos para el corazón
El balón o globo de contrapulsación aórtica Arrow es un dispositivo que, conectado a un catéter, se coloca en la aorta para reducir la carga de trabajo del corazón. Es fabricado por Arrow International, una división de Teleflex Medical.
El 11 de marzo de 2016, la FDA emitió una alerta mundial que sigue vigente, advirtiendo que más de 20 lotes del Arrow IAB de 30 cc entrañaban un riesgo para la vida de los pacientes. La Anmat había autorizado su importación en 2012 y la renovó en 2013 y 2018.
American Fiure importó siete modelos de este dispositivo en 2015, otros 7 en 2016 y 9 en 2017, con la alerta vigente. “No coincidieron los lotes que tuvieron el problema”, aseguraron desde la Anmat.
Sin embargo, desde American Fiure reconocieron que efectuaron un retiro del mercado: “Por pedido de nuestro proveedor del exterior retiramos del mercado las unidades que habíamos distribuido hasta el momento y las despachamos de vuelta a la fábrica de origen. No tuvimos reportes de eventos con pacientes, entendemos que todos los productos fueron recuperados y devueltos al fabricante”.
Otro globo de contrapulsación intraaórtica, en este caso marca Datascope, tuvo una alerta sanitaria de la FDA en julio de 2017 que sigue vigente. Este dispositivo se coloca en la aorta para mejorar el aporte de oxígeno y reducir la carga de trabajo del corazón en casos de infarto. Lo fabrica Datascope y en la Argentina lo importa la empresa Debene y la Fundación Favaloro.
La alerta fue emitida por el riesgo de muerte para los modelos CS100 y sugirió verificaciones para el modelo CS300. Este último ya había ingresado a la Argentina siete veces entre marzo y diciembre de 2017, y el primer modelo, con un mayor riesgo potencial, había sido importado con anterioridad a la alerta. Desde la Anmat reconocieron que “hubo una acción de campo” pero agregaron que “la información es confidencial”.
La Fundación Favaloro sí respondió con más detalle a la consulta y reveló que la importadora -Devene- nunca cumplió con lo informado a la Anmat. “La Institución cuenta con dos consolas CS100. Fuimos notificados de esto por la empresa Debene el 28 de noviembre de 2017 –(es decir, varios meses después de que se conociera el alerta mundial) pero hasta la fecha no han recibido la pieza de reemplazo preventivo”. Los equipos nunca se corrigieron para reparar la posible falla. “De nuestra parte dejamos indicada la situación en el equipo para que se tomen los recaudos en caso de ocurrencia. Como aclaración, nunca ocurrió esta falla potencial”, señalaron.
Otro equipo para pacientes con insuficiencia cardíaca – el dispositivo de asistencia ventricular (VAD) Heartware– también fue comercializado en el mercado argentino pese a que, entre 2013 y 2018, hubo al menos 13 alertas de la FDA por peligro de muerte. Durante estos años, el producto ingresó al país importado por Medikar SA. Se trata de una bomba mecánica que sustituye a las bombas naturales del corazón cuando éstas fallan.
“Hubo un problema con el pin, que se podía doblar. La acción de campo era advertir que, de no ser colocado correctamente, el pin se podía doblar. Se reforzaron las instrucciones de uso”, admitió la Anmat.
Sin embargo, la importadora Medikar aseguró que si bien ingresaron al país 6 unidades del dispositivo “los números de serie no están comprendidos en la alerta”, y que “4 de ellas fueron utilizadas y 2 permanecen en stock en la empresa”. En su respuesta, agregó: “Nuestra compañía es informada por el fabricante siempre que corresponda de cada una de las acciones de campo llevadas a cabo en relación al producto HVAD. Respecto a este recall en particular aún no hemos recibido comunicación, entendemos que se debe a que no nos involucra”.
Sistema de localización intraoperatorio BrainLab
Un dispositivo que permite a los cirujanos hacer incisiones en el cráneo del paciente con mayor precisión, conocido como sistema de localización intraoperatorio BrainLab, también fue incluido en una alerta de la FDA en enero del 2016 y se extendió por dos años. Dos de estos dispositivos utilizados en neurocirugía fueron importados el 20 de noviembre de 2016, con la alerta mundial vigente.
Como requieren que el médico ingrese datos de manera digital, un error en el software del dispositivo como el reportado “podría intensificar pequeñas imprecisiones” en las incisiones durante la intervención.
“Hubo acción de campo. Se hizo una actualización del software”, justificó la Anmat. Desde AADEE no respondieron en forma particular y solo señalaron: “En términos generales cuando el proveedor avisa que hay un problema con el software se avisa a los clientes. La Casa matriz nos envía el nuevo software y los técnicos corrigen los equipos”.
Asistencia para respirar
La marca CareFusion advirtió un riesgo potencial por la falla de un componente del ventilador AVEA, un dispositivo médico que proporciona ventilación asistida a pacientes infantiles con insuficiencia respiratoria. La FDA emitió la alerta el 3 abril de 2015, que sigue vigente. En la Argentina, la Anmat había autorizado la inscripción del producto a la empresa Driplan SA en septiembre de 2010. El permiso fue revalidado en febrero de 2013 y en marzo de 2016.
De acuerdo con los registros aduaneros, al país ingresaron al menos 6 ventiladores Carefusion AVEA entre marzo y diciembre de 2015, otros dos en febrero de 2016 y tres más durante 2017. O sea que la Anmat autorizó su ingreso al país cuando ya tenía una advertencia de riesgo Clase 1. Desde el organismo, se limitaron a decir que la empresa “efectuó una acción de campo mediante el cambio de las plaquetas del ventilador”.
La importadora Driplan SA dio su versión de los hechos: “Los equipos no tenían que retirarse del mercado, podían continuar funcionando sin problema. CareFusion lanzó una acción voluntaria correctiva sobre estos equipos, sobre un lote, y decía que había que reemplazar un componente. Nos mandaron una nota, informamos a Anmat y procedimos a la acción correctiva que era el cambio del componente”. Consultados sobre qué instituciones médicas tenían los equipos y cuantos equipos fueron, señalaron: “Nosotros tenemos un contrato de confidencialidad con la fábrica y no se pueden brindar detalles sobre los equipos en particular”.
El respirador Newport HT70, un dispositivo de asistencia ventilatoria de Covidien, del grupo Medtronic, comenzó a reportar fallas en abril de 2013. Los usuarios advirtieron problemas en las baterías de algunos dispositivos. La FDA publicó varios recall Clase 1 sobre este equipo, en abril de 2013 y marzo 2015. Luego existieron otras alertas nuevas sobre el mismo producto. En la Argentina, la ANMAT lo autorizó en octubre de 2010 a la importadora Age Medical SA. Al menos cinco respiradores fueron importados durante 2011.
Desde la importadora reconocieron que “los equipos fueron importados y vendidos por Age Medical” pero que “al momento del recall, ya no era representante de Newport dado que Covidien había adquirido la fábrica y tenía oficinas en Argentina”. Desde Age Medical agregaron: “Dado el conocimiento que nosotros teníamos de los clientes y equipos, Covidien contrató nuestro soporte técnico para hacer el cambio de las baterías, pero fueron ellos los responsables de la acción”.
El ventilador Respironics V60 de Philips, destinado a auxiliar a pacientes con insuficiencia respiratoria, registró inconvenientes en algunas series de fabricación en junio de 2013, debido a un problema con el software en el ensamblaje de la placa de administración de energía.
La FDA emitió una alerta el 4 de junio de 2013, ya que tenía un potencial riesgo de vida para los pacientes. El 24 de abril de 2017 la FDA emitió un nuevo aviso, ya que otros lotes del mismo producto reportaron fallas eléctricas. La ANMAT autorizó su importación a fines de diciembre de 2010 y en septiembre de 2011.
Según los registros aduaneros, se importaron al menos cuatro respiradores entre 2013 y 2014, cuando ya regía la alerta, y otro más en 2016 por la propia Phillips. Desde la ANMAT se limitaron a informar que “existió una acción de campo” para corregir el problema en la Argentina, pero no brindó más detalles.
Philips respondió a la consulta que “en 2013 y 2017 procedió al retiro voluntario del mercado” de este modelo de ventilador. “En ambos retiros voluntarios, Philips notificó debidamente a clientes, distribuidores y agencias reguladoras de todo el mundo. No se recibió información sobre ningún evento adverso en Argentina”, aseguró.
Esta empresa tuvo 19.686 efectos adversos reportados en 2017 ante la FDA, según el análisis realizado por ICIJ en base a datos de las autoridades reguladoras de los Estados Unidos.
El equipo argentino del Consorcio Internacional de Periodismo de Investigación que participó de The Implant Files está integrado por Mariel Fitz Patrick (Infobae); Maia Jastreblansky, Iván Ruiz y Ricardo Brom (La Nación); Emilia Delfino (Perfil) y Sandra Crucianelli (para Perfil).