Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
El Congreso de EE UU está evaluando la reautorización de los programas que permiten que la FDA cobre tarifas por la revisión de los medicamentos de venta con receta (Prescription Drug User Fee Act o PDUFA), genéricos (Generic Drug User Fee Amendments GDUFA), y biosilimares (BsUFA). Como parte de este proceso, el 12 de enero de 2022, la FDA entregó un documento al comité de salud, que es parte del Comité de Energía y Comercio, en el que relata todas sus actividades desde la última reautorización y sus planes para el futuro.
A continuación, resumimos el testimonio de Patricia Cavazzoni y Peter Marks [1], funcionarios de la FDA, frente al Comité del Congreso, y el de la Dra. Reshma Ramachandran investigadora y miembro del National Clinician Scholars Program at Yale School of Medicine. [2]. Ambos documentos son de acceso libre. El primero tiene como objetivo resumir el informe de la FDA sobre los tres programas mencionados y lograr su reautorización por lo que ofrece una imagen muy positiva de los que la FDA ha logrado con esos fondos. La Dra. Ramachandran sintetiza los avances de la FDA, pero señala sus deficiencias, la necesidad de que el Congreso apoye a la FDA para que no tenga que depender de las industrias que debe regular, y señala algunas iniciativas que contribuirían al fortalecimiento de la agencia.
Ambos documentos incluyen muchas referencias a documentos de interés.
Testimonio de Patrizia Cavazzoni y Peter Marks
PDUFA. La FDA espera trabajar con el Congreso en la reautorización de la PDUFA para seguir acelerando el desarrollo y la aprobación de medicamentos y productos biológicos vitales que sean seguros y eficaces.
La revisión oportuna de la seguridad y eficacia de las solicitudes de nuevos medicamentos (NDA en inglés new drug application???) y de las solicitudes de licencia de productos biológicos (BLA biological license application) es fundamental para que la FDA pueda cumplir su misión de proteger y promover la salud pública, y el PDUFA es esencial para estos esfuerzos. En concreto, el PDUFA autoriza a la FDA a recaudar tarifas de usuario a las industrias para, entre otras cosas, contratar personal adicional y gestionar y mejorar los sistemas de información. Las tarifas de usuario recaudadas en virtud del PDUFA han permitido acelerar el proceso de revisión de las solicitudes de nuevos medicamentos y productos biológicos sin comprometer los elevados estándares de la FDA en cuanto a la seguridad, eficacia y calidad de los nuevos medicamentos.
A través de las sucesivas reautorizaciones de PDUFA, el programa ha ido evolucionando; y ahora incluye un amplio proceso de comunicación y consulta entre los patrocinadores de medicamentos y la FDA a lo largo de todo su proceso de desarrollo. Esto nos permite proporcionar más orientación a los patrocinadores, establecer expectativas más claras sobre los datos necesarios para que la agencia pueda revisar y evaluar adecuadamente un medicamento, lo cual mejora la posibilidad de que sea aprobado durante el primer ciclo y permite el acceso más rápido de los pacientes a los nuevos productos.
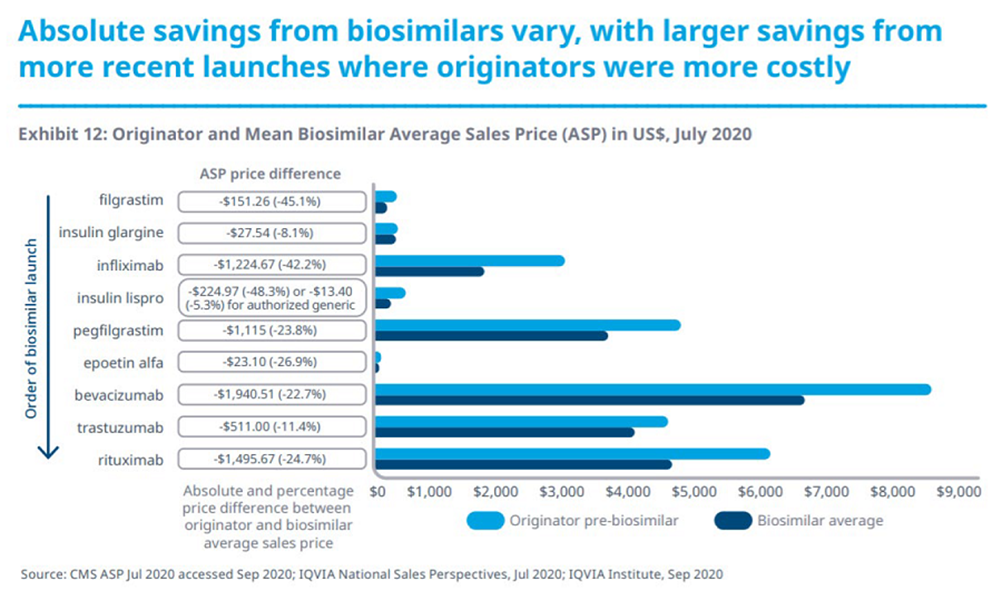
Como se muestra en la Figura 1, hoy en día, casi dos tercios de las nuevas sustancias activas que se comercializan en todo el mundo se lanzan primero en EE UU.
Durante la evolución de este programa, la FDA ha revisado muchas solicitudes y ha cumplido con el cronograma establecido por PDUFA (Ver Figura 2). Esto se logró incluso cuando aumentaron mucho las solicitudes de revisión durante la pandemia por covid 19.
Un elemento clave del éxito de PDUFA es la interacción continua con los patrocinadores de medicamentos. Esto se refleja en el aumento del número de programas de desarrollo de medicamentos en curso (IND en inglés investigational new drug) y aumento de solicitudes de las empresas para reunirse con personal de la FDA, como se muestra en la Figura 3.
La mejora de la calidad de los programas de desarrollo de fármacos y de las solicitudes presentadas, como resultado de las interacciones con la FDA explica que el 87% de las solicitudes de comercialización de NDAs y BLAs se aprobarán durante el primer ciclo de revisión en el 2021.
Muchos medicamentos y productos biológicos que se someten a revisión prioritaria también se benefician de otros programas destinados a acelerar su desarrollo, como la designación de vía rápida (fast track) y de producto innovador (breakthrough). Ambos programas ofrecen mayores oportunidades de interacción entre los patrocinadores y los revisores de la FDA a lo largo del proceso de desarrollo, incluyendo la asesoría de la FDA sobre el diseño y la realización de los ensayos clínicos necesarios para demostrar la seguridad y la eficacia del producto. Además, la designación de innovación suele incluir una mayor participación de la dirección de la FDA.
La aprobación acelerada, otro programa de revisión rápida, también acelera el proceso de desarrollo al acortar los ensayos clínicos previos a la comercialización. La aprobación acelerada acepta la utilización de criterios de valoración indirectos o subrogados que tienen una probabilidad razonable de predecir el beneficio clínico, y se aprueban con el requisito de confirmar su beneficio clínico durante el periodo de post comercialización. Treinta y nueve de las 60 aprobaciones de NME de 2021 (65%) utilizaron uno o más programas acelerados, específicamente la designación de vía rápida, la designación de innovación y/o la aprobación acelerada.
Figura 1. Proporción de nuevas sustancias activas comercializadas por región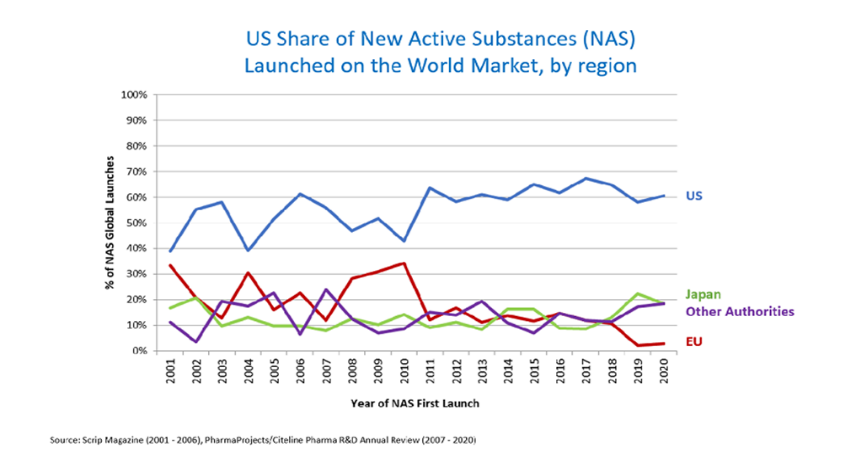
Figura 2. Proporción de revisiones concluidas dentro del plazo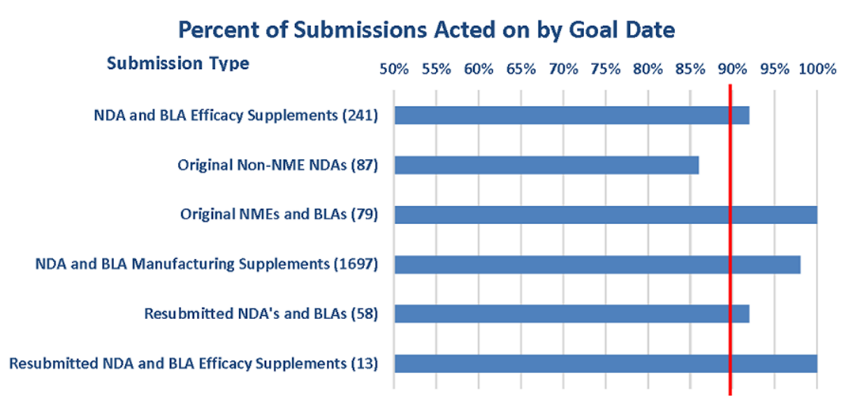
Figura 3: Solicitudes de reunión, y IND activas
Figura 4: aprobaciones de nuevos medicamentos y biológicos según mecanismo de aprobación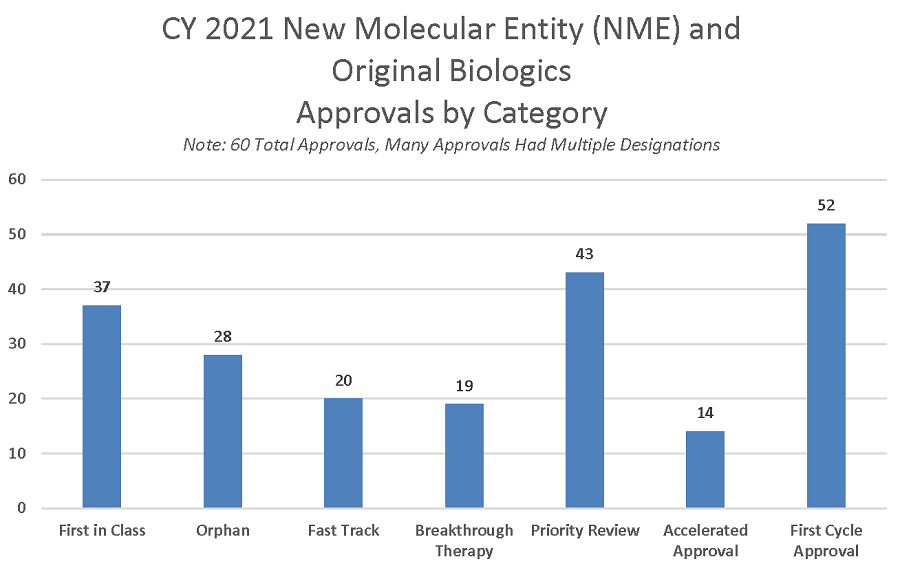
En marzo de 2020, la FDA anunció la creación de un programa para la revisión y el desarrollo de posibles terapias para el covid-19, el Programa de Aceleración del Tratamiento del Coronavirus, o “CTAP”. Su objetivo principal es acelerar el desarrollo de terapias para pacientes y consumidores. En el marco del CTAP, la FDA ha utilizado toda la autoridad disponible y la flexibilidad normativa adecuada para facilitar el desarrollo de productos seguros y eficaces para tratar a los pacientes con covid-19. A 31 de diciembre de 2021, había más de 670 programas de desarrollo de fármacos en fase de planificación y revisamos más de 470 ensayos de posibles terapias para covid-19, incluyendo antivirales, inmunomoduladores, anticuerpos neutralizantes, terapias celulares y genéticas, y combinaciones de estos productos.
Reautorización PDUFA VII. De acuerdo con el mandato del Congreso, la FDA ha realizado reuniones públicas, negociaciones con la industria regulada y consultas periódicas con las partes interesadas, incluidos los defensores de los pacientes y los consumidores, para determinar las prioridades para PDUFA VII.
Las recomendaciones para el PDUFA VII se centran en garantizar que la FDA tenga capacidad para revisar productos nuevos e innovadores, incluyendo los productos de terapia celular y genética. A tal fin, PDUFA VII recomienda aumentar las tarifas para financiar 352 posiciones de tiempo completo nuevas y para apoyar las inversiones críticas en la infraestructura del programa, como la modernización de los sistemas de información.
GDUFA. El programa GDUFA ha contribuido a ampliar el acceso de los pacientes a los medicamentos genéricos de buena calidad. Según la Asociación de Medicamentos Accesibles, a partir de un análisis de IQVIA, entre 2022 y 2020 los medicamentos genéricos han ahorrado al sistema de salud estadounidense US$2,4 billones.
Ha habido un aumento constante de solicitudes de procesos posteriores a la aprobación de las solicitudes abreviadas de nuevos medicamentos genéricos (ANDA), incluyendo los Suplementos de Aprobación Previa y las presentaciones de Cambios Efectuados (CBE) (Figura 5). La mayoría de ellas se refieren a cambios en las plantas de manufactura o en el etiquetado/ficha técnica. Además, con la aprobación constante de nuevas entidades moleculares y segundos usos también habrá un aumento en las solicitudes de aprobación de productos genéricos (ANDA).
GDUFA II, permitió que la FDA se pusiera al día con cientos de ANDA. Se aprobaron más de 3.000 ANDA y, se emitieron más de 50.000 comunicaciones a la industria para facilitar el desarrollo de genéricos. En el marco del GDUFA II, la Agencia se comprometió a evaluar el 90% de las ANDA prioritarias en los ocho meses siguientes a su presentación, y a evaluar el 90% de las ANDA estándar en diez meses. El programa ha superado sus objetivos (Figura 6).
La revisión prioritaria está disponible para las solicitudes de medicamentos genéricos con competencia limitada, así como para los medicamentos genéricos que escasean y cumplen ciertas condiciones. Esto incluye un plazo de revisión más corto bajo el marco de correspondencia previa sobre la instalación (PFC), bajo el cual los patrocinadores presentan información sobre las plantas de manufactura y las pruebas del medicamento a más tardar 60 días antes de la presentación de la solicitud.
Un elemento central es acelerar la evaluación de los ANDA de posibles “primeros genéricos” porque pueden abrir el mercado a la competencia de los genéricos por primera vez. Muchas ANDA de “primer genérico” no pueden presentarse legalmente hasta una fecha específica después de la aprobación del medicamento innovador. La figura 7 muestra el número de aprobaciones de primeros medicamentos genéricos por año fiscal durante la GDUFA II.
Figura 5: Solicitudes de procesos post aprobación de ANDA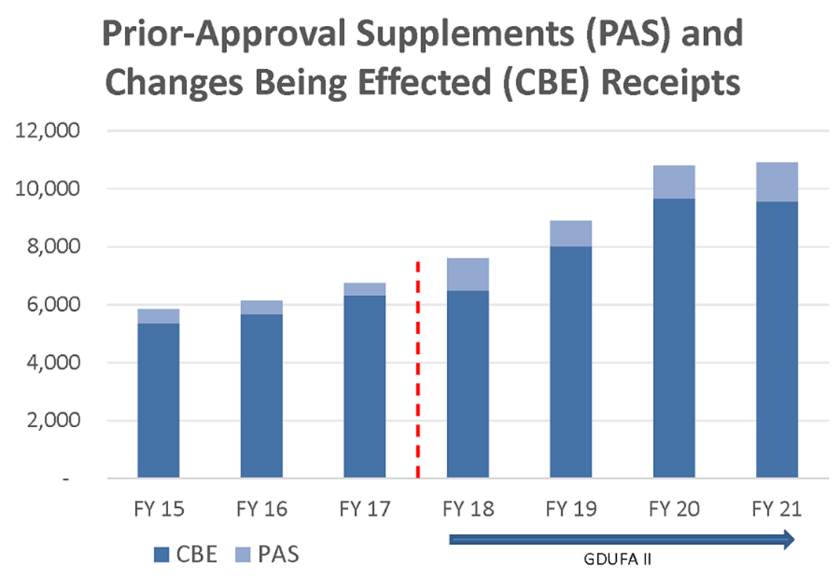
Figura 6: Cumplimiento del cronograma de revisión de ANDA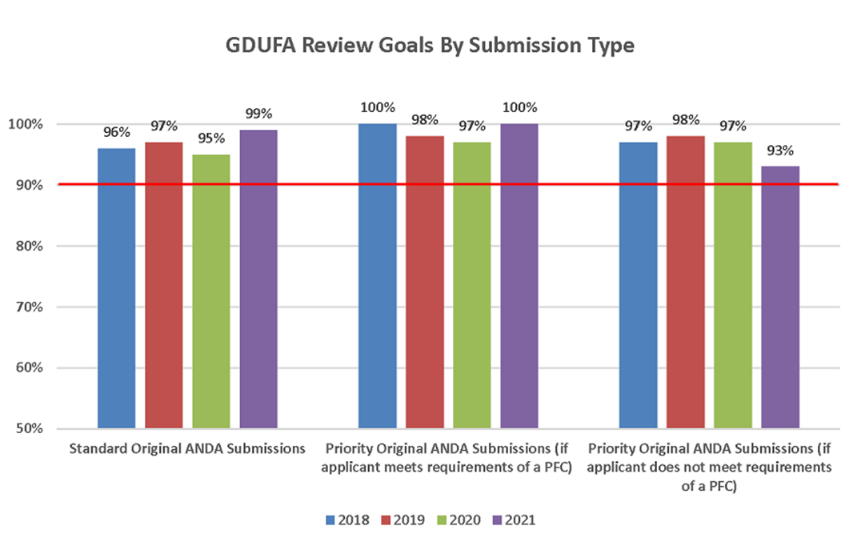
Figura 7: Aprobaciones de primeros productos genéricos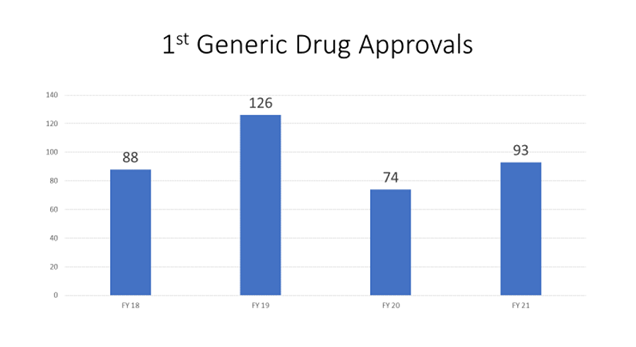
Programa-Pre -ANDA. Para reducir el número de ciclos hasta la aprobación, especialmente en el caso de los productos genéricos complejos, se estableció un programa de pre-ANDA, que ayuda a clarificar las expectativas regulatorias, permitiendo que los solicitantes presenten solicitudes más completas. Este programa incluye mecanismos de consulta a la FDA en relación con estos productos después de la presentación de la ANDA.
Para los productos complejos, este programa estableció tres tipos de reuniones (donde la FDA provee asesoría, reuniones previas a la presentación del ANDA para explicar el contenido y el formato del ANDA, y la reunión donde la FDA informa de las posibles deficiencias de la solicitud y se planifican los siguientes pasos).
En noviembre 2020, la FDA publicó la guía “Formal Meetings Between FDA and ANDA Applicants of Complex Products Under GDUFA Guidance for Industry”. Para facilitar el desarrollo de nuevos productos genéricos, la FDA emitió unas Guías Específicas de Producto (PSG). El 15 de enero de 2022, había 1.949 PSG disponibles.
La FDA consulta y solicita las aportaciones del público, la industria y los investigadores académicos para desarrollar una lista anual de iniciativas científicas y reguladoras específicas para la investigación de los medicamentos genéricos. Además, involucra a las partes interesadas a través de numerosos talleres científicos y publica un informe anual sobre los logros.
Mejoras en el programa de evaluación de ANDA. Cuando se presenta una ANDA, la FDA determina en primer lugar si la ANDA está lo suficientemente completa como para poder hacer una evaluación sustantiva. Mientras se está evaluando el ANDA, la FDA comunica las deficiencias que ha encontrado durante la evaluación aproximadamente a partir de la mitad del proceso de revisión. Las comunicaciones continúan de forma continua durante la evaluación.
Cuando las deficiencias de una ANDA impiden la aprobación de la FDA, ésta emite una carta de respuesta completa en la que se detallan las deficiencias que se deben corregir para que sea aprobada, y después se pueden establecer teleconferencias para orientar a los patrocinadores.
Mejoras en el programa de evaluación de archivos maestros de medicamentos (DMF en inglés drug master files). Los DMF de tipo II son presentaciones de un tercero, distinto del solicitante de la ANDA, que contienen información confidencial sobre un principio activo (o ingrediente farmacéutico activo [API]) o un producto intermedio (o los materiales utilizados en su preparación) que la Agencia evalúa de forma independiente. Varios productores genéricos pueden utilizar los mimos DMFs. Es esencial que haya buena comunicación entre los titulares de los DMFs, los solicitantes del ANDA y la FDA, para reducir la posibilidad de que surjan problemas y se atrasen las aprobaciones, y la FDA lo ha facilitado estandardizando las cartas.
La FDA publicó un borrador de guía sobre los cambios posteriores a la aprobación de un DMF de tipo II de API y los mecanismos de presentación de solicitudes ANDA que hacen referencia a un DMF de tipo II de API. La FDA también publicó un borrador de guía titulado “Completeness Assessments for Type II API DMFs under GDUFA” (Evaluaciones de integridad de los DMF de tipo II de las API en virtud del GDUFA).
Mejoras en la evaluación de las instalaciones. Para mitigar los problemas con la exportación que habían identificado los fabricantes de API con sede en EE UU, se solicitó que la FDA que emitiera guías y realizara actividades de divulgación para los reguladores extranjeros sobre su modelo de selección de centros de fabricación basado en el riesgo. La FDA publicó un manual de políticas y procedimientos (MAPP) titulado “Understanding CDER’s Risk-Based Site Selection Model” en 2018, para explicar cómo la FDA determina qué instalaciones de fabricación deben priorizarse para las inspecciones de vigilancia de rutina.
La FDA mejoró la velocidad y la transparencia de las comunicaciones relativas a los resultados de las inspecciones de las instalaciones, y publica estos resultados en su página web.
Mejoras en la rendición de cuentas y la presentación de informes. La agencia desarrolló un programa para documentar los avances hacia alcanzar los objetivos de GDUFA II. También mejoró la transparencia y eficiencia en la administración, la asignación y el uso de los recursos generados a través de las tarifas de usuario. Esa información está en la página web bajo “Enhanced Accountability & Reporting“.
La FDA publicó un Plan Financiero Quinquenal en el año fiscal 2018, con actualizaciones anuales, y celebró una reunión anual sobre la transparencia financiera y la eficiencia de los programas de tarifas de usuario. También se contrató a un externo para que evaluara la metodología para ajustar los presupuestos de acuerdo a la carga de trabajo (CPA), tanto para PDUFA, como para GDUFA y BsUFA.
La reautorización de GDUFA III. Alrededor del 30% de los productos de referencia activos, que no tienen competencia genérica, son productos complejos. Por lo tanto, las negociaciones de la GDUFA III se centraron en aprovechar los éxitos de la GDUFA II, proponiendo nuevos procesos y procedimientos para lograr aprobaciones en ciclos más tempranos y mejorando el programa pre-ANDA.
Evaluaciones de ANDA. La carta de compromiso de GDUFA III propone minimizar la emisión de cartas de respuesta completa y mejorar la correspondencia sobre las plantas de manufactura previas al ANDA para informar la decisión de la FDA sobre la necesidad de hacer una inspección. Se pretende mejorar la oportunidad de las respuestas reglamentaria ampliado el uso de correspondencia y teleconferencias.
Archivos maestros de medicamentos e instalaciones de fabricación. Ampliar las oportunidades de evaluación temprana de los DMFs antes de que se presenten ciertas ANDA prioritarias y entre los ciclos de revisión, para aumentar la probabilidad de que el DMF sea adecuado al mismo tiempo que la ANDA asociada. Revisar los plazos de respuesta a la correspondencia.
La mayoría de las instalaciones cumplen los requisitos de las buenas prácticas de fabricación vigentes. Para ayudar a los que no las cumplen, las instalaciones para fabricar medicamentos genéricos elegibles podrían solicitar una reunión posterior a la carta de advertencia para compartir con la FDA información preliminar sobre la adecuación y la integridad de sus planes de acción correctiva.
Programa previo a la ANDA y ciencia reglamentaria. Seguir mejorando la ciencia reglamentaria y agilizar el desarrollo de medicamentos genéricos complejos mediante la introducción de mejoras adicionales en los programas. Establecer objetivos para los PSGs de medicamentos complejos. Mejorar la transparencia entorno a los nuevos PSGs y las revisiones de PSGs. Ampliar las reuniones y mejorarlas para que la FDA pueda proveer asesoría científica para que los titulares del ANDA puedan solventar las deficiencias.
Contratación. Serecomienda contratar a 128 trabajadores de tiempo completo.
Mejorar la gestión de los recursos procedentes de las tarifas de los usuarios. Mejorar la agilidad operativa del programa GDUFA y la gestión de los recursos de las tarifas de los usuarios a través de mejoras en la capacidad para planear el uso de los recursos y para estimar las necesidades futuras. Se ha hecho una propuesta para que los legisladores autoricen un aumento de los recursos (de hasta el 3% por encima de la inflación) para poder responder a las necesidades que vayan surgiendo según las cargas de trabajo.
Esta propuesta legislativa también eliminaría el ajuste reglamentario del último año y lo sustituiría por un ajuste de la reserva de funcionamiento, para proporcionar a la Agencia la opción de aumentar los ingresos para ayudar a garantizar los recursos adecuados en el caso de una importante falta de recaudación de tarifas u otras interrupciones en la financiación. Este ajuste de la reserva de funcionamiento permitiría que la agencia aumentara las tasas para mantener una reserva de funcionamiento de entre 8 y 10 semanas de tarifas de usuario. Si las reservas de funcionamiento previstas superan las 12 semanas de gastos de funcionamiento, la FDA estaría obligada a reducir las tarifas de ese año fiscal para que la reserva de funcionamiento no supere las 12 semanas de tarifas.
El GDUFA III mantendría la transparencia financiera mediante la publicación de un plan financiero quinquenal y la celebración de una reunión pública para debatir el plan y otros compromisos financieros cada año fiscal.
BsUFA. Los productos biológicos han contribuido a la salud de muchos pacientes, incluyendo a los pacientes con cáncer, artritis reumatoidea y diabetes. Los biosimilares pueden mejorar el acceso y ahorrar costos a los pacientes y financiadores de los servicios de salud.
Ley de Innovación y Competencia de Precios de Productos Biológicos de 2009 (Biologics Price Competition and Innovation Act of 2009 BPCI).Esta ley estableció una vía de aprobación abreviada para los productos biológicos que demuestren que son “biosimilares” o “intercambiables” con el biológico de referencia autorizado por la FDA. Un producto biosimilar es muy parecido al producto de referencia, a pesar de las pequeñas diferencias en los componentes clínicamente inactivos, sin diferencias clínicamente significativas en términos de seguridad, pureza y potencia. Un producto intercambiable es un producto biosimilar que cumple el requisito adicional de demostrar que el producto produce el mismo resultado clínico que el producto de referencia en cualquier paciente y, para un producto biológico que se administra más de una vez a un paciente, el riesgo en términos de seguridad o de disminución de la eficacia de alternar o cambiar entre el producto biosimilar y el de referencia no es mayor que el riesgo de usar el producto de referencia sin dicha alternancia o cambio. En algunos estados, los farmacéuticos pueden sustituir un producto biológico por un biosimilar intercambiable sin consultar con el prescriptor.
La vía de aprobación abreviada permite que una solicitud de biosimilar se base, en parte, en la determinación previa de la FDA de que el producto de referencia es seguro y eficaz, lo que ahorra al solicitante tiempo y recursos y, por tanto, fomenta la competencia.
La FDA aprobó el primer biosimilar, Zarxio, el 6 de marzo de 2015, bajo la Ley de Reautorización de la FDA de 2017 (FDARA). Cuando se promulgó la BsUFA II, solo había cinco productos biosimilares aprobados para cuatro productos de referencia. Durante la BsUFA II el número ha crecido a 33 biosimilares para 11 productos de referencia, incluyendo dos biosimilares intercambiables a partir del 19 de enero de 2022.
Un análisis reciente de IQVIA proporciona datos sobre el ahorro potencial con los biosimilares.
La BsUFA II ha permitido la implementación de un nuevo modelo de revisión y mayor comunicación con las empresas, lo que ha facilitado el desarrollo de productos biosimilares. Este nuevo modelo de revisión promueve la eficiencia y eficacia del proceso de revisión durante el primer ciclo, y tiene como objetivo minimizar el número de ciclos de revisión que serán necesarios para la aprobación. Las mejoras en el intercambio de información entre la industria y la FDA han contribuido a aumentar las aprobaciones durante el primer ciclo, alcanzando casi el 70% en comparación con el 39% durante la BsUFA I.
La FDA publicó un borrador de guía “Formal Meetings Between the FDA and Sponsors or Applicants of BsUFA Products” y una guía final “Best Practices for Communication Between IND Sponsors and FDA During Drug Development”. El 3 de enero de 2022, había cerca de 100 programas de desarrollo de biosimilares activos, y se habían recibido solicitudes de reuniones para discutir el desarrollo de biosimilares para otros 47 productos de referencia. Todo esto con el objetivo de que la comunicación sea más clara, concisa y oportuna.
También se invirtió en capacitar al personal y mejorar los sistemas de contratación. la FDA se comprometió a hacer una evaluación completa y continua de las prácticas de contratación y retención. Esto ha permitido que la FDA elabore todas las guías a las que se había comprometido bajo el BsUFA II
La BsUFA II incluía objetivos relacionados con la publicación de información sobre productos biológicos. En este sentido, en 2020, la FDA publicó “The Purple Book: Base de datos de productos biológicos autorizados” que es de acceso público e incluye información importante sobre los biológicos, incluyendo información sobre las presentaciones del producto, la potencia y las formas de dosificación, además de otros campos de datos. Esta base de datos cuanta con una herramienta de búsqueda.
La FDA ha mejorado la gestión de los recursos del programa y la capacidad para anticipar las necesidades a más largo plazo, incluyendo un plan financiero quinquenal con actualizaciones anuales, informes financieros anuales y reuniones públicas para hablar de las finanzas del programa.
Reautorización de BSUFA III (años fiscales 2023-2027). El plan de la agencia ha sido elaborado tras consultar con la industria, expertos científicos y académicos, profesionales de la salud, defensores de pacientes y consumidores. Todas las actas de las reuniones son de acceso público.
Mejora de los procesos de revisión previa a la comercialización, los procedimientos y el rendimiento. La agencia espera recibir solicitudes suplementarias para los biosimilares ya aprobados. Por ello, la BsUFA III propone nuevas categorías de suplementos, plazos de revisión y objetivos de rendimiento para acelerar la revisión, incluyendo la revisión de las actualizaciones de seguridad en los etiquetados/fichas técnicas. Además, esta propuesta modifica algunas de las reuniones formales para mejorar la comunicación y la retroalimentación durante el proceso de desarrollo/
Mejora del desarrollo de productos biosimilares e intercambiables y de la ciencia reguladora. Para ello se incorporarán las mejores prácticas en la comunicación entre la FDA y los patrocinadores a través de actualizaciones de las guías pertinentes, el Manual de Políticas y Procedimientos (MAPP) y la Política y Procedimientos Operativos Estándar (SOPP). Esto incluye:
Para avanzar aún más en el desarrollo de productos biosimilares intercambiables seguros y eficaces, la BsUFA III propone emitir cuatro guías, y organizar talleres científicos.
También se establecería un programa piloto de ciencia regulatoria, que se centraría en dos proyectos de demostración: (1) avanzar en el desarrollo de productos intercambiables, y (2) mejorar la eficiencia del desarrollo de productos biosimilares.
Mejoras continuas para la gestión de los recursos de las tasas de usuario. Al igual que PDUFA y GDUFA.
Contrataciones. La agencia querría contratar a 15 personas de tiempo completo, y mejorar el sistema de contratación y retención de personal.
Modernización y transparencia en los sistemas informáticos de información.
Conclusión. Las tarifas de usuario son fundamentales para garantizar que la FDA disponga de los recursos necesarios para llevar a cabo las revisiones de forma oportuna sin comprometer los elevados estándares de la Agencia, todo ello para conseguir que los productos médicos seguros y eficaces lleguen antes a los pacientes. La reautorización de PDUFA, GDUFA y BsUFA permitirá a la FDA aprovechar el éxito demostrado por los programas, beneficiando aún más a los pacientes y afirmando la posición de nuestra nación como líder mundial en innovación biomédica.
Testimonio de Reshma Ramachandran
Reshma Ramachandran [2] ofreció un testimonio escrito, en el que habla de la importancia tener una FDA fuerte, menciona algunos aspectos de los acuerdos de tarifas de usuario que son esenciales para mejorar la salud de los pacientes, y discute las oportunidades que tiene el Congreso de apoyar y proteger a una agencia que es vital para la salud pública de nuestra nación. A nuestro parecer es una propuesta que fortalecería a la FDA y que se debería tener en cuenta, aunque como ella misma reconoce al final, parece que no ha captado el interés de la FDA y habrá que ver si influye en las posiciones de los congresistas.
Aquí el resumen de su testimonio
Cuando la FDA concede un permiso de comercialización se entiende que, antes de determinar si estos nuevos tratamientos son realmente, seguros y eficaces, la agencia ha revisado los datos de ensayos clínicos bien diseñados. También se asume que la FDA ha certificado, tras las debidas diligencias, que los procesos de fabricación de estos medicamentos son seguros y de buena calidad.
Al igual que muchos de mis colegas, me preocupa que la creciente dependencia de las tarifas a los usuarios, provenientes de las industrias que la FDA regula, haya influido indebidamente en el marco legal y regulatorio, convirtiéndose en una agencia demasiado laxa.
La agencia ha acelerado la aprobación de medicamentos y productos biológicos, y esto se ha asociado a ensayos clínicos menos sólidos y mayor incertidumbre sobre el beneficio clínico de los tratamientos. Para que la FDA mantenga su independencia como autoridad reguladora nacional, el Congreso debería asignar más fondos a la agencia, reduciendo así su dependencia de la industria y mitigando su indebida influencia, pues no prioriza las necesidades de los pacientes. No obstante, dada la falta de fondos suficientes, el Congreso debería intervenir para garantizar que estos acuerdos de tarifas de usuario incluyan salvaguardias para proteger a los pacientes y la salud pública.
Es importante que la FDA cuente con el personal necesario para evaluar la eficacia y seguridad de los medicamentos, y agilizar el proceso de revisión de medicamentos genéricos e intercambiables.
La FDA se ha comprometido a acercarse gradual y reflexivamente a la generación de evidencia utilizando datos de la práctica clínica y diseños complejos de ensayos clínicos. La FDA tiene que ser cuidadosa al evaluar si tales métodos no sólo son más eficientes, sino también suficientes para tomar decisiones regulatorias.
La FDA también se ha comprometido a reforzar la vigilancia de la seguridad de los medicamentos, incluyendo la Iniciativa Centinela. Esto ayudará a evaluar las señales de seguridad, de modo que la agencia pueda tomar medidas más oportunas y prevenir los efectos adversos relacionados con los medicamentos.
Los acuerdos sobre las tasas de usuario ofrecen varias oportunidades para que el Congreso refuerce aún más el proceso de revisión de la FDA y se centre más en el paciente, con el fin de garantizar el acceso oportuno a los fármacos y medicamentos biológicos que han demostrado ser realmente eficaces y seguros. Esto podría lograrse a través de las siguientes iniciativas.
Para medir el desempeño de la FDA, utilizar medidas más significativas desde el punto de vista clínico que estén centradas en el paciente. Como parte de los acuerdos sobre las tarifas de usuarios, la FDA ha propuesto una serie de objetivos de rendimiento y medidas relacionadas con la revisión oportuna y la respuesta a las solicitudes regulatorias relacionadas con medicamentos y biológicos. Aunque es importante mejorar la eficiencia, la agencia también debería medir su rendimiento en términos de aportes a la clínica y a los pacientes. Por ejemplo, podría informar:
Revisión de los estándares de evidencia que se requieren para los medicamentos y productos biológicos que se aprueban a través de las vías de revisión acelerada. Las vías de revisión acelerada, como la aprobación acelerada, han permitido que los pacientes accedan antes a los medicamentos que han demostrado efectividad en base a criterios de valoración indirectos (subrogados) en lugar de criterios clínicos, y eso ha generado incertidumbre sobre su beneficio clínico. En el marco de la aprobación acelerada, la FDA exige la realización de estudios adicionales después de su aprobación para confirmar el hipotético beneficio clínico de los medicamentos y biológicos en un plazo determinado. Una vez finalizados estos estudios, la FDA revisa los datos de los ensayos confirmatorios para determinar si estos medicamentos pueden optar a la aprobación estándar.
Sin embargo, un análisis realizado por la revista British Medical Journal encontró que de 253 fármacos aprobados a través de la vía de aprobación acelerada entre 1992 y 2020 casi la mitad (n=112) no habían confirmado ser clínicamente eficaces; y una quinta parte de ellos (n=24) habían estado en el mercado durante más de cinco años. Sólo dieciséis de todos los medicamentos que habían recibido la aprobación acelerada durante este periodo fueron retirados, bien por falta de eficacia o porque los ensayos confirmatorios nunca se llevaron a cabo.
En el caso del aducanumab, la FDA ha concedido a Biogen un plazo de nueve años para completar sus ensayos confirmatorios, que es mucho más largo que la duración media de los ensayos postcomercialización o el plazo de notificación de resultados para otros medicamentos aprobados por la misma vía. Muchos estudios post comercialización se atrasan y la FDA ha renunciado a los ensayos posteriores a la aprobación para algunos medicamentos. Es más, cuando estos ensayos confirmatorios requeridos se llevan a cabo y muestran resultados negativos, la FDA no ha retirado el permiso de comercialización para esas indicaciones.
Por último, algunos de los ensayos clínicos postcomercialización siguen utilizando criterios de valoración indirectos, a pesar de que el objetivo de estos ensayos es confirmar beneficio clínico (esto ocurrió para el 40% de los oncológicos aprobados por la FDA entre 1992 y 2017). Es más, algunos de estos estudios no están bien diseñados (muestras pequeñas, sin aleatorización, abiertos).
Como parte de los acuerdos de tarifas de usuario, la FDA se debería comprometer a revisar los estándares de evidencia para la aprobación a través de vías rápidas, como la aprobación acelerada. La FDA no debería permitir que las aprobaciones aceleradas se mantengan a perpetuidad. La FDA podría retirar las indicaciones de aprobación acelerada si los ensayos confirmatorios posteriores a la aprobación se retrasan significativamente o no muestran un beneficio clínico.
Las autoridades reguladoras de otros países, como el Reino Unido, Australia y Europa, que conceden aprobaciones rápidas, exigen su renovación cada uno o dos años. Como parte del proceso de renovación, los patrocinadores deben presentar un informe provisional sobre cualquier obligación pendiente, incluyendo los plazos para la realización de ensayos confirmatorios. En cualquier caso, si los ensayos confirmatorios arrojaran resultados negativos, la FDA también debería retirar automáticamente la aprobación acelerada.
La FDA debería informar a los pacientes y a los médicos de que se ha concedido la aprobación acelerada para un nuevo medicamento, y que el medicamento podría ser retirado si los ensayos confirmatorios no aportan un beneficio clínico para los pacientes.
Además, la FDA debería exigir que los ensayos confirmatorios de los medicamentos que han recibido la aprobación acelerada utilicen criterios de valoración clínicos y estén bien diseñados y ejecutados.
Uso adecuado y transparente de la evidencia de la práctica clínica para la toma de decisiones regulatorias. La FDA ha estado evaluando la calidad y la aceptabilidad de la evidencia de la práctica clínica (RWE) para la toma de decisiones regulatorias según lo dispuesto en la Ley de Curas del Siglo 21, en particular para apoyar la aprobación de nuevas indicaciones de los medicamentos aprobados o para satisfacer los requisitos de los estudios de postcomercialización. También han surgido propuestas legislativas para permitir el uso de datos de la práctica clínica (RWD), incluyendo los registros de pacientes, las historias clínicas electrónicas, las facturas médicas y la tecnología digital para cumplir con los requisitos de los estudios postcomercialización. La FDA se ha comprometido a realizar un plan piloto para estudiar el uso de estos datos en las decisiones regulatorias.
Investigaciones recientes han planteado dudas sobre la viabilidad de utilizar los RWD para respaldar determinadas decisiones regulatorias. Un estudio descubrió que solo el 15% de los ensayos clínicos realizados en EE UU y publicados en revistas médicas de alto impacto podrían replicarse de manera factible utilizando los datos de facturas o historias clínicas electrónicas. Otro estudio analizo si fuera factible utilizar dichos datos para emular los ensayos confirmatorios postcomercialización requeridos por la FDA para los medicamentos que recibieron aprobación acelerada entre 2009 y 2018. De los 50 ensayos confirmatorios requeridos por la FDA, ninguno podría emularse de manera factible utilizando los RWD disponibles. Esto sugiere que es poco probable que los métodos observacionales y de los RWD actualmente disponibles sustituyan a los requisitos de ensayos confirmatorios postcomercialización, como se ha propuesto.
Los RWD no permiten controlar adecuadamente los criterios de inclusión y exclusión, ni los criterios de valoración que se requieren en los estudios. La agencia debería invertir en más investigación para delinear claramente las condiciones de uso apropiadas de RWD en la toma de decisiones regulatorias, y esto se debería hacer antes de que haya un mandato legal que le impida determinar donde se ha demostrado que los RWD beneficia a los pacientes.
Los estudios de RWE deberían estar disponibles en un registro público.
Garantizar la finalización oportuna y la integridad de los estudios postcomercialización. Un estudio descubrió que de los requisitos y compromisos de postcomercialización emitidos por la FDA en 2009 y 2010, casi el 20% ni siquiera se habían iniciado, el 25% se habían retrasado o estaban en curso, y poco más del 50% se habían completado entre cinco y seis años después de la aprobación del medicamento. En un estudio de los medicamentos aprobados por la vía acelerada entre 2009 y 2013, solo dos quintas partes tenían pruebas confirmatorias de eficacia en los tres años posteriores a la aprobación.
Los tiempos para que se completen estos estudios varían ampliamente y suelen ser más largos de lo requerido.
Mantenimiento de normas rigurosas para la aprobación de terapias para enfermedades con necesidades insatisfechas
Terapias celulares y genéticas. Un estudio analizó las cinco nuevas terapias génicas que habían recibido la aprobación de la FDA a 31 de diciembre de 2020. A través de la designación de terapia avanzada de medicina regenerativa (en inglés RMAT), la FDA ha podido acelerar con éxito el desarrollo y la entrada en el mercado de estos productos. Sin embargo, esta rapidez también parece haberse logrado a expensas de la calidad de la información disponible antes de la comercialización. La mayoría de las autorizaciones de estas terapias génicas se basaron en un pequeño ensayo abierto, de un solo brazo, que utilizó un marcador indirecto como criterio de valoración primario de su eficacia. Además, la FDA sólo exigió estudios post comercialización para tres de los cinco tratamientos.
Dado que estos productos se aprueban con evidencia limitada, la FDA debería exigir estudios de postcomercialización y recopilación de datos continuos sobre estos pacientes, a través de registros.
Antibióticos y antifúngicos. Los incentivos financieros para desarrollar productos para enfermedades infecciosas están mal alineados, y recompensan la comercialización de productos con una eficacia poco clara contra los patógenos resistentes. En lugar de conceder costosos incentivos financieros por tratamientos no probados, la FDA debería exigir a los patrocinadores de los candidatos a fármacos antimicrobianos que realicen ensayos clínicos pivotales en poblaciones de pacientes clínicamente relevantes, incluyendo las que suelen estar excluidas, y que estos ensayos clínicos estén diseñados para demostrar superioridad frente a alternativas conocidas y eficaces.
Mantener los requisitos de inspección de las instalaciones de fabricación in situ. Estas inspecciones son fundamentales para garantizar que los productos médicos son seguros, de alta calidad y eficaces para los pacientes. No se deben sustituir con modalidades de inspección remota.
Mantener la responsabilidad legal de la FDA de exigir que se publiquen los resultados de los ensayos clínicos. Los patrocinadores de los ensayos clínicos tienen la obligación de registrar y, posteriormente, incluir la información de resultados de los ensayos clínicos en ClinicalTrials.gov, generalmente en un plazo de 12 meses a partir de la fecha de finalización del ensayo. La transparencia de dicha información de resultados es importante, ya que informa las decisiones de atención médica y los futuros esfuerzos de investigación.
A través de una investigación en curso, utilizando la Ley de Libertad de Información, obtuvimos todas las medidas coercitivas que la FDA ha tomado por el incumplimiento de estos requisitos hasta finales de abril de 2021. Todas ellas fueron en forma de Avisos Preliminares de Incumplimiento (o Preavisos), de los cuales la FDA había emitido sólo 58 para los patrocinadores de los ensayos. Ninguno de estos preavisos se dirigió a organismos federales, incluyendo los Institutos Nacionales de Salud (NIH), a pesar de que los NIH son los patrocinadores de muchos ensayos que no han presentado la información de sus resultados a ClinicalTrials.gov según lo exigido. También descubrimos que estos preavisos eran extremadamente eficaces para mejorar el cumplimiento: a mediados de agosto del año pasado, más del 90% de los destinatarios habían comunicado la información que faltaba a ClinicalTrials.gov en un plazo medio de tres semanas.
Está claro que la FDA podría y debería enviar más preavisos y avisos y dar a conocer públicamente estos esfuerzos.
Aumentar la transparencia general de los procesos de toma de decisiones regulatorias. La agencia se ha comprometido a trabajar más estrechamente con la industria, y ha dicho que organizaría talleres públicos. Es necesario que la agencia se comprometa a solicitar y garantizar la participación de partes interesadas independientes.
Aprendiendo de la experiencia con el aducanunab, la FDA debería instituir un cortafuegos entre el personal que ayuda a la industria a preparar las solicitudes regulatorias y el que realiza la revisión. De este modo, los pacientes y los médicos estarán más seguros de que la agencia mantiene su propia independencia, sin la influencia indebida de los patrocinadores de medicamentos en la toma de decisiones regulatorias.
Al revisar la carta de compromiso del PDUFA VII divulgada por la FDA a finales de septiembre del año pasado y el proyecto de ley recientemente publicado, las disposiciones parecen ser casi idénticas a pesar de los comentarios de organizaciones independientes de defensa de los pacientes y de consumidores que solicitaron revisiones específicas a los acuerdos propuestos. La FDA, en su respuesta a estos comentarios públicos, declaró que “no ha introducido cambios en esas recomendaciones para la reautorización del PDUFA” porque existe “un apoyo general al acuerdo PDUFA VII”. Teniendo en cuenta esta respuesta, y el hecho de que la FDA mantiene negociaciones separadas y no transparentes con las empresas biofarmacéuticas y grupos comerciales, parecería que la agencia está dando más peso a las recomendaciones de la industria que a las de las organizaciones que representan de forma independiente los intereses del público. Convocar a todas las partes interesadas en las futuras negociaciones de acuerdos de tarifas de usuario sería un primer paso para restaurar el equilibrio en la asimetría para influir en el proceso redactar las cartas de compromiso de la FDA para el programa de tarifas de usuario y la legislación correspondiente.
En estos acuerdos de tarifas de usuario, el Congreso y la FDA deben considerar tanto los beneficios como los riesgos, tanto a corto como a largo plazo, de las propuestas incluidas. Para mí está claro que, en general, los beneficios de contar con una FDA adecuadamente financiada superan cualquier riesgo potencial. Sin embargo, hay que hacer más reformas para garantizar que la FDA cumpla su mandato y su promesa de proteger y mejorar la salud de nuestros pacientes y de nuestra nación.
Referencias