Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
Cochrane es una red global independiente compuesta por profesionales de la salud, investigadores, defensores de pacientes y otros, que enfrenta el desafío de lograr que las enormes cantidades de evidencia generadas a través de la investigación sirvan para sustentar las decisiones en materia de salud. Cochrane es una organización sin ánimo de lucro con colaboradores de más de 120 países que trabajan juntos para producir información fiable y accesible sobre la salud, libre de patrocinios comerciales y de otros conflictos de interés.
La Colaboración para la Integridad y la Transparencia de la Investigación (The Collaboration for Research Integrity and Transparency o CRIT por sus siglas en inglés) es una iniciativa interdisciplinaria lanzada en 2016 en la Universidad de Yale para mejorar la calidad y la transparencia de la investigación relacionada con productos médicos. Por medio de la investigación, la abogacía en defensa de la integridad de la ciencia y el litigio, CRIT busca garantizar que la evidencia clínica que respalda y fundamenta nuestra comprensión de la seguridad y eficacia de los productos farmacéuticos, los dispositivos y otros insumos médicos sea exacta, completa, accesible y confiable.
Transparencia Internacional (Transparency International o TI) es la principal organización no gubernamental del mundo dedicada a la lucha contra la corrupción. Con más de 100 capítulos a nivel mundial, TI cuenta con una amplia experiencia y conocimiento de la corrupción a nivel global. El Programa de Productos Farmacéuticos y Salud (PHP por sus siglas en inglés) es una iniciativa global con sede en Transparencia Internacional del Reino Unido. El objetivo general del programa es mejorar la salud mundial y los resultados de la atención médica en beneficio de todas las personas, de todas las edades. Se propone conseguirlo reduciendo la corrupción y promoviendo la transparencia, la integridad y la responsabilidad en los sectores farmacéutico y de la salud.
TranspariMED es una iniciativa que busca acabar con la distorsión de la evidencia en la medicina, y lo hace desarrollando y promoviendo políticas para mejorar la transparencia en los ensayos clínicos.
El presente estudio fue elaborado por Till Bruckner, fundador de TranspariMED, en estrecha colaboración con los miembros del equipo de Cochrane, el CRIT y el Programa de Productos Farmacéuticos y Salud de Transparencia Internacional.
Si bien el contenido de este estudio es responsabilidad exclusiva de Cochrane, CRIT, el Programa Farmacéutico y de Salud de Transparencia Internacional y TranspariMED, muchos expertos externos aportaron generosamente su tiempo para revisar y dar su opinión sobre los primeros borradores del documento. Queremos agradecer a las siguientes personas por sus valiosas contribuciones:
Erick Turner (Universidad de la Salud y las Ciencias de Oregón), Simon Kolstoe (Universidad de Portsmouth), Vaseeharan Sathiyamoorthy (Organización Mundial de la Salud), Fahmy Aboulenein-Djamshidian (Universidad Médica de Viena, TI-Austria), Yannis Natsis (Alianza Europea de Salud Pública), Stuart Buck (Fundación Laura y John Arnold), Al Weigel (Sociedad Internacional de Profesionales de Publicaciones Médicas) y otros tres expertos que solicitaron mantener el anonimato.
Autor: Till Bruckner (TranspariMED) – tillbruckner@gmail.com
Editora: Sarah Harris-Steingrüber (TI-PHP)
Diseño: Jon Le Marquand (TI-UK)
©2017 Transparencia Internacional Reino Unido. Todos los derechos reservados. Este estudio se publica bajo una licencia Creative Commons BY 3.0. Cualquiera puede citar, reproducir o reutilizar parte o la totalidad de su contenido sin permiso previo, siempre y cuando se cite el estudio original o se establezca un enlace al mismo.
Publicado en diciembre de 2017.
ISBN: 978-1-910778-75-3
Se ha hecho todo lo posible para verificar la exactitud de la información contenida en este informe. Toda la información se considera correcta hasta diciembre de 2017. No obstante, Transparencia Internacional Reino Unido no puede aceptar la responsabilidad de las consecuencias de su uso para otros fines o en otros contextos.
El número de organización benéfica registrada de Transparencia Internacional del Reino Unido es 1112842
Resumen Ejecutivo
El problema
Los ensayos clínicos son un motor clave de la innovación y el progreso en la medicina, pero los científicos saben desde hace décadas que la base de evidencia existente sobre medicamentos y dispositivos médicos es incompleta y está sesgada debido a la opacidad de los ensayos clínicos. Tanto la comunidad médica como el sector privado y los organismos públicos carecen de acceso a información fiable sobre los beneficios y daños de los medicamentos, dispositivos y tratamientos. Las consecuencias negativas de esta falta de transparencia son graves:
Esta falta de transparencia en los ensayos clínicos puede aumentar el riesgo de influencia indebida, manipulación de los datos y distorsión de la evidencia. Es un síntoma de la limitada intervención de la autoridad reguladora en el proceso de informar. Abre la puerta al fraude y a la corrupción y socava tanto los avances médicos como los objetivos de salud pública.
La solución
Una mayor transparencia en los ensayos clínicos beneficiaría de manera positiva y directa los resultados en los pacientes, mejoraría la asignación de los escasos recursos para la investigación médica y los servicios de salud, y facilitaría y aceleraría el desarrollo de nuevos tratamientos y curas. La transparencia de los ensayos clínicos se basa en cinco pilares diferentes:
Implementación
Naciones Unidas ha solicitado recientemente a los gobiernos que tomen medidas para resolver este acuciante problema de salud pública. Lograr que los ensayos clínicos sean más transparentes sería una intervención de bajo costo y altamente rentable. De hecho, con los marcos legales actuales sólo con medidas administrativas se pueden lograr importantes avances en materia de transparencia.
Medidas concretas
Los responsables de la toma de decisiones deberían adoptar las tres medidas siguientes para aumentar la transparencia de los ensayos clínicos y lograr que el sector responda mejor a los ciudadanos, los pacientes, los contribuyentes y los inversionistas:
Primera medida: Garantizar que los ensayos clínicos financiados con fondos públicos se publiquen de manera transparente
Como un primer paso, los responsables a nivel político deberían exigir que todos los organismos públicos que financian investigación dentro de su jurisdicción adopten y amplíen las normas de transparencia de la Organización Mundial de la Salud para la divulgación de los resultados de los ensayos clínicos, y que se aseguren de su plena aplicación. Con este sencillo primer paso se obtendrían importantes beneficios a un costo mínimo.
Segunda medida: Hacer cumplir las normas existentes para la presentación de informes sobre los ensayos clínicos
En segundo lugar, los responsables de la toma de decisiones deben proporcionar a los organismos gubernamentales los recursos, las competencias y el apoyo político que necesitan para hacer cumplir las leyes, las normas y los reglamentos existentes, que actualmente no se suelen aplicar de forma coherente. Los responsables de la toma de decisiones deben apoyar a las agencias gubernamentales en el desarrollo de mecanismos eficaces de control que acarreen sanciones para lograr que el sector rinda cuentas.
Tercera medida: Reforzar los marcos legales y regulatorios.
En tercer lugar, los responsables de la toma de decisiones deben adaptar las leyes, normas y reglamentos existentes a los estándares mundiales de mejores prácticas y garantizar que todos los ensayos clínicos, pasados y presentes, respeten los cinco pilares de la transparencia en los ensayos clínicos.
ANTECEDENTES
¿Qué son los ensayos clínicos?
Los ensayos clínicos son un motor clave de la innovación y el progreso en la medicina. Los investigadores médicos reclutan voluntarios para participar en ensayos con el fin de investigar si los medicamentos, dispositivos médicos y tratamiento son seguros y eficaces [1]. Los ensayos clínicos suelen determinar la eficacia de un fármaco, dispositivo o tratamiento administrándolo a una serie de pacientes comparando los resultados con un grupo de control que recibe otro fármaco o un placebo. Los científicos hacen el seguimiento de los participantes en el ensayo, en ambos grupos, para detectar cambios en su estado de salud, así como cualquier efecto secundario negativo que puedan presentar. Al analizar los datos y comparar los dos grupos, los investigadores comprueban si un fármaco, dispositivo o tratamiento es seguro y eficaz.
El proceso de investigación y desarrollo médico es complejo, largo y costoso. Cada año, las empresas farmacéuticas, las universidades y otros grupos de investigación realizan alrededor de 20.000 ensayos clínicos en los que participan más de dos millones de pacientes en todo el mundo, con un costo estimado de más de US$60.000 millones [2]. Los ensayos de mayor envergadura pueden involucrar a múltiples financiadores, numerosas instituciones de investigación y miles de pacientes en varios países diferentes, lo que presenta formidables desafíos regulatorios.
El diseño, la realización y los resultados de los ensayos clínicos, así como la forma de comunicarlos, tienen importantes repercusiones comerciales y de salud pública. Los organismos gubernamentales utilizan los resultados de los ensayos clínicos para decidir si permiten la comercialización de un nuevo medicamento o dispositivo y si financian su suministro. Igualmente, los ensayos clínicos sirven de base para que los médicos que procuran determinar las mejores opciones de tratamiento para sus pacientes tomen sus decisiones.
¿Por qué son tan importantes los ensayos clínicos?
Los ensayos clínicos son el fundamento de la medicina basada en la evidencia. Los organismos reguladores del gobierno, las agencias de salud pública, las aseguradoras y los médicos de familia se basan en los resultados de los ensayos clínicos para tomar decisiones médicas que pueden salvar vidas. Buscando en los registros de ensayos clínicos y revisando los resultados de los mismos, pueden ver lo que los investigadores de todo el mundo han descubierto y decidir cuáles son las mejores opciones de tratamiento.
Además, los registros de ensayos aportan una visión general del estado actual de los conocimientos médicos y de los proyectos de investigación en curso, permitiendo que los financiadores y a los científicos eviten duplicaciones inútiles y aprovechen el trabajo de los demás (Zarin et al., 2008).
Ensayos no confiables: sesgo al informar y distorsión de la evidencia
Los científicos saben desde hace décadas que la base de evidencia que existe sobre los medicamentos y dispositivos médicos puede estar sesgada. Numerosos estudios han demostrado que la evidencia que se publica sobre medicamentos y dispositivos médicos exagera sistemáticamente los beneficios y resta importancia a los daños (Bekelman et al., 2003; Goldacre, 2012; Golder et al., 2016; Sani, 2014; Song et al., 2010).
Dos factores importantes que contribuyen a este problema son el sesgo al informar y la distorsión de la evidencia.
El sesgo al informar se produce cuando es más probable que se divulguen los resultados de los ensayos éxitosos que los de los ensayos que fracasan. El sesgo al informar se debe a múltiples causas. Los actores comerciales tienden a dar prioridad a la publicación de evidencia que hace que sus productos parezcan buenos [3], y a veces utilizan mecanismos contractuales o presiones informales para impedir que los científicos publiquen los resultados de ensayos que van en contra de sus intereses comerciales (Angell, 2005; Bass, 2008; Lexchin, 2003; Steinbrook, 2005).
Incluso cuando no hay intereses comerciales en juego, puede haber sesgos (McGauran et al., 2010). Los editores de las revistas académicas suelen preferir publicar los resultados positivos de ensayos que podrían presagiar nuevos avances médicos. Los científicos lo saben y pueden no intentar publicar los resultados de ensayos con resultados nulos o negativos; y si lo intentan, posiblemente no encuentren una revista de alto perfil dispuesta a publicar su artículo (Song et al., 2014).
|
Cómo un importante ensayo clínico estuvo a punto de convertirse en despilfarro en investigación En 2015, el Dr. Aus Alzaid, un médico en ejercicio que trabajaba en Arabia Saudí, se propuso descubrir si un medicamento para la diabetes ampliamente utilizado por millones de pacientes en todo el mundo (entre ellos, algunos de los suyos) podía afectar la memoria o causar demencia. Descubrió que la evidencia disponibles públicamente sobre la posible relación del fármaco con la demencia eran contradictorias y se limitaban a datos de laboratorio y encuestas de observación. Sólo existía un único ensayo clínico relevante. Según su inscripción en el registro, se había completado tres años antes, pero sus resultados no se habían compartido públicamente de ninguna forma, exponiendo a millones de pacientes a daños no reconocidos. Los resultados no se publicaron hasta que el Dr. Alzaid y otros lo solicitaron reiteradamente. Si el ensayo no se hubiera registrado, habría permanecido completamente invisible, y sus resultados se habrían perdido para siempre. El Dr. Alzaid comentó posteriormente que “no debe quedar en manos de un médico al azar o de un particular reclamar los resultados de los ensayos clínicos o rogar personalmente a los investigadores que publiquen su trabajo una vez concluido. La publicación de los ensayos clínicos registrados es una responsabilidad profesional, no una prerrogativa personal del investigador principal. Esto se establece claramente en todos los códigos de conducta profesionales” (Aizaid, 2016). |
Los ensayos que no se han registrado ni publicado permanecen completamente invisibles. Esto es un grave problema para los científicos, incluso para los que trabajan en las agencias reguladoras nacionales [4], que necesitan ver todos los ensayos realizados hasta la fecha para poder determinar si un fármaco o dispositivo es seguro y eficaz [5]. Los ensayos invisibles provocan un considerable despilfarro en la investigación: los descubrimientos valiosos no se comparten y los científicos pueden explorar una y otra vez los mismos callejones sin salida.
La distorsión de la evidencia al revelar los resultados de los ensayos es la segunda razón por la cual la evidencia publicada sobre medicamentos y dispositivos médicos exagera los beneficios y minimiza los daños. Esta distorsión toma muchas formas, como la manipulación estadística, la comunicación selectiva de resultados parciales y (con mucha menos frecuencia) la manipulación directa de los datos [6]. Aunque todas las formas de distorsión de la evidencia se consideran antiéticas, y muchas se clasifican como mala conducta científica, muy pocas constituyen casos de corrupción flagrante o delitos penales. Aunque algunas formas de distorsión de pruebas están muy extendidas, rara vez se detecta a los autores y es poco probable que lleguen a sufrir consecuencias.
La distorsión de la evidencia obedece a una serie de factores, desde el sesgo de confirmación [7] hasta las ambiciones profesionales de los científicos o los intereses financieros. En algunos casos, científicos con escasos conocimientos estadísticos pueden distorsionar la evidencia involuntariamente, sin darse cuenta de que están generando datos engañosos, violando las normas científicas y éticas, y pudiendo causar daños a los pacientes.
|
Cuantificando el sesgo de publicación y la distorsión de la evidencia Un equipo de investigadores analizó los informes de los ensayos clínicos de 12 antidepresivos aprobados por la FDA de EE UU. El equipo localizó 74 ensayos registrados por la FDA en los que participaron 12.564 pacientes y comparó las evaluaciones realizadas por los expertos de esa agencia con la bibliografía académica disponible. Todos menos uno de los 38 ensayos clínicos que la FDA calificó de positivos fueron publicados. De los 36 ensayos restantes con resultados negativos, sólo se publicaron tres. Los 33 ensayos restantes no se habían publicado en absoluto (sesgo de publicación, 22 ensayos), o se habían publicado de tal forma que se sugería que los resultados habían sido positivos (distorsión de la evidencia, 11 ensayos). Así, mientras los expertos de la FDA habían llegado a la conclusión de que casi la mitad de los 74 ensayos no habían tenido un resultado positivo, los médicos e investigadores que se basaban únicamente en la literatura académica habrían tenido la impresión de que la gran mayoría (49 de 52) de todos los ensayos relevantes sobre los antidepresivos habían obtenido un resultado positivo (Turner et al., 2008). En 2015, otro análisis de 15 medicamentos aprobados por la FDA descubrió que, por cada medicamento, alrededor del 35% de los resultados de todos los ensayos clínicos realizados para obtener la aprobación de esta agencia no estaban disponibles para los médicos ni para los investigadores externos (Miller, 2015). |
El precio de la opacidad en los ensayos clínicos
En medicina, el sesgo al informar y la distorsión de la evidencia han estado fuera de control durante décadas debido a la opacidad selectiva de los ensayos clínicos. Los pacientes, los médicos y las agencias de salud pública, al no poder analizar cómo se generaron esos hallazgos, se ven obligados a confiar en los resultados prefabricados de la investigación que presentan las entidades que tienen interés en exagerar los beneficios de los medicamentos y dispositivos médicos y en restar importancia a sus daños.
Entre las consecuencias negativas de esta falta de transparencia están el daño directo a los pacientes, la ralentización del progreso científico y el aumento del riesgo financiero para los inversionistas.
Los pacientes se ven perjudicados. La falta de transparencia en los ensayos clínicos perjudica a los pacientes. Los beneficios y riesgos de los fármacos y dispositivos médicos no se pueden entender y evaluar plenamente si la información que se genera sobre ellos a través de la investigación es escasa, sesgada, está distorsionada o es incompleta. Hay numerosos casos bien documentados en los que un gran número de pacientes se han visto afectados por la escasa transparencia de los ensayos clínicos.
Las agencias de salud pública no pueden tomar decisiones con conocimiento de causa. Cuando las empresas farmacéuticas solicitan una licencia para comercializar un nuevo fármaco o dispositivo médico, entregan a las agencias reguladoras, como la FDA de EE UU y la Agencia Europea de Medicamentos (European Medicines Agency, EMA), una gran cantidad de información generada durante los ensayos clínicos. Sin embargo, tanto las empresas como los organismos reguladores suelen negarse a compartir esa información con terceros. En consecuencia, los investigadores independientes y otras agencias gubernamentales [8]no pueden revisar o volver a analizar los datos presentados por los actores comerciales.
Varios medicamentos y dispositivos aprobados por los organismos reguladores han sido retirados del mercado posteriormente por motivos de seguridad cuando los pacientes experimentaron efectos secundarios nocivos inesperados (Onakpoya, 2016). En consecuencia, los científicos que trabajan en las agencias reguladoras y otros organismos de salud pública suelen ser firmes defensores de una mayor transparencia en los ensayos clínicos.
Se desperdician los fondos para la salud pública. Debido al rápido aumento de los costos de la atención en salud a nivel mundial, los sistemas de salud pública y las aseguradoras privadas se enfrentan a decisiones difíciles sobre qué tratamientos financiar. Para determinar si la eficacia de un medicamento justifica su costo, los responsables de la toma de decisiones necesitan tener acceso a los resultados completos de todos los ensayos clínicos. Sin embargo, estos datos no suelen estar disponibles. En el pasado, esto ha provocado el despilfarro de cuantiosos fondos públicos.
Se ralentiza el progreso médico. La escasa transparencia de los ensayos clínicos impide que los financiadores de la investigación y los científicos coordinen eficazmente sus esfuerzos. Se calcula que cada año se malgastan US$85.000 millones en la financiación de investigación médica, ya que ensayos que cuestan millones no contribuyen al progreso médico porque sus resultados no se revelan [9]. Los científicos repiten innecesariamente ensayos de medicamentos que otros ya han descubierto que son perjudiciales o ineficaces, o ambas cosas. Todo esto retrasa el desarrollo de nuevos tratamientos y curas, y socava la preparación para las emergencias de salud pública, como las epidemias.
Los accionistas se exponen a riesgos considerables. Los inversionistas de las empresas farmacéuticas se han sumado a la lucha por una mayor transparencia en los ensayos clínicos, porque la falta de transparencia los expone a riesgos sustanciales de mercado, legales y regulatorios (AllTrials, 2015; The Economist, 2015). La asimetría de información entre las empresas y los inversionistas debilita la eficiencia del mercado de capitales. Los inversionistas no pueden evaluar de manera fiable el potencial de mercado de los nuevos medicamentos en desarrollo, ni determinar si hay datos ocultos cuya aparición podría amenazar los flujos de ingresos existentes o dar lugar a acciones legales (Feuerstein, 2016).
De la opacidad a la transparencia en los ensayos clínicos
En la actualidad, los ensayos clínicos se caracterizan por un alto nivel de opacidad (CRIT, 2017). Tanto la comunidad médica como el sector privado y los organismos públicos carecen de acceso a información fiable sobre los beneficios y los perjuicios de los medicamentos, dispositivos y tratamientos. Esto es una falta de ética, y la asimetría de información resultante afecta negativamente la salud de las personas, la salud pública, las finanzas públicas y el funcionamiento eficaz de los mercados.
En el resto de este documento se expondrán las medidas concretas que pueden adoptar los responsables de la toma de decisiones para que el ámbito de la investigación médica clínica se ajuste a las normas mundiales de transparencia.

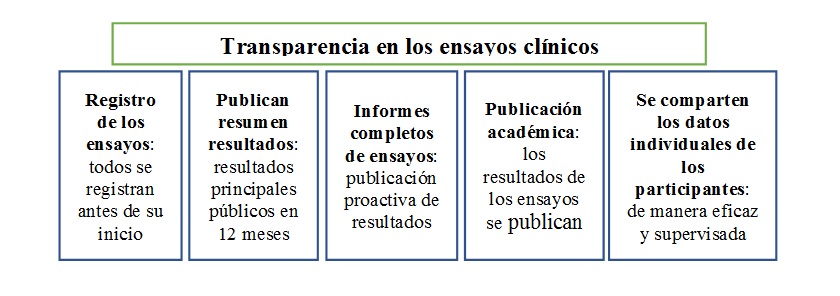
Los cinco pilares de la transparencia en los ensayos clínicos
El fortalecimiento de la transparencia en los ensayos clínicos beneficiaría positiva y directamente la salud de los pacientes, mejoraría la asignación de los escasos recursos para investigación médica y para la atención en salud, y facilitaría y aceleraría el desarrollo de tratamientos y curas nuevos y eficaces (Bruckner y Ellis, 2017). La transparencia en los ensayos clínicos se sustenta en cinco pilares distintos, a saber: el registro anticipado de los ensayos, la publicación oportuna de un resumen de los resultados en el mismo registro, la divulgación de los informes completos de los ensayos, la publicación imparcial y precisa de los resultados de los ensayos en revistas académicas y el intercambio de datos de los participantes individuales.
Registro de los ensayos. El registro prospectivo de los ensayos en una base de datos regulada en línea está reconocido universalmente como una obligación ética, independientemente de los requisitos legales nacionales. Reduce la posibilidad de sesgo y distorsión de la evidencia en la difusión de los resultados de los ensayos [10]. Permite que los financiadores de las investigaciones eviten la duplicación de investigaciones previas e identifiquen legítimas lagunas del conocimiento, y ayuda a los científicos a aprovechar los descubrimientos realizados por otros. En muchas jurisdicciones, el registro de los ensayos ya es un requisito legal o regulatorio, pero su cumplimiento sigue siendo irregular.
Publicación de resúmenes de los resultados. Una vez finalizado un ensayo clínico, los investigadores están obligados a publicar el resumen de sus resultados en el registro (o registros) en que se inscribió el ensayo originalmente [11]. Esto proporciona al público un panorama instantáneo de los resultados principales del ensayo. La publicación de los resultados permite a los científicos compartir rápida y sistemáticamente los nuevos descubrimientos sin tener que esperar a la publicación académica, que puede tardar varios años, y reduce el potencial de sesgo y distorsión de la evidencia en la comunicación de los resultados. Algunos reglamentos y leyes ya exigen que las instituciones de investigación publiquen el resumen de los resultados de algunos ensayos en un plazo de 6 a 12 meses tras su conclusión [12], pero su cumplimiento es deficiente.
Informes completos de los ensayos. El componente principal de los informes completos de los ensayos son los Informes de Estudios Clínicos (IEC o Clinical Study Reports -CSRs), que son documentos extensos que permiten que los expertos determinen la importancia y la fiabilidad de los resultados de un ensayo y señalar los beneficios o efectos secundarios adicionales que el equipo que originalmente realizó la investigación puede haber pasado por alto. Esto favorece el progreso médico al tiempo que reduce la posibilidad de distorsión de la evidencia y el fraude en la comunicación de los resultados. Las empresas farmacéuticas ya están obligadas a proporcionar a las autoridades reguladoras los informes de estudios clínicos cuando solicitan la licencia para comercializar un medicamento, pero estos informes no se suelen poner a disposición de terceros.
|
Resúmenes de resultados versus Informes de Estudios Clínicos (IEC o CSR) Los resúmenes de resultados adoptan la forma de un breve cuadro que resume las características clave de un ensayo clínico: el fármaco o dispositivo investigado, las medidas de resultado utilizadas, las características de los pacientes y los principales hallazgos. Los resúmenes de resultados permiten acceder públicamente a un panorama general de los hallazgos de un ensayo, pero no contienen información en profundidad sobre el diseño, la realización o los resultados del ensayo. En cambio, los informes de estudios clínicos (IEC o CSR , el componente principal de los informes completos de los ensayos) son documentos muy técnicos que suelen tener más de mil páginas y ofrecen una imagen muy detallada del diseño, la realización, el análisis y los hallazgos de un ensayo, incluyendo detalles sobre los efectos secundarios negativos que experimentaron los pacientes [13]. |
Publicación académica. Las revistas académicas son la principal plataforma de comunicación para muchos científicos. A menudo son el primer recurso que tienen los médicos cuando buscan información sobre cómo tratar a sus pacientes, y lo más importante es que son revisadas críticamente por otros científicos o clínicos en un proceso llamado revisión por pares. Los resultados de los ensayos deberían publicarse en revistas o ponerse a disposición del público para garantizar que los descubrimientos de los ensayos clínicos se comparten ampliamente e influyan en la práctica médica. En la actualidad, muchos de los resultados de los ensayos no se publican en las revistas.
Compartir los datos de los participantes individuales. Compartir los datos sobre cada participante individual (DPI) que se han recogido durante el ensayo clínico puede acelerar el progreso médico, aumentar la comprensión de la seguridad y la eficacia de los medicamentos, dispositivos y tratamientos y reducir las posibilidades de distorsión y fraude en la presentación de la evidencia. Sin embargo, todavía existen importantes obstáculos prácticos para compartir los DPI de forma efectiva. Aunque numerosas iniciativas están intentando superar estos problemas, solo se podrá aprovechar plenamente el inmenso potencial de compartir los DPI si los otros pilares de la transparencia de los ensayos clínicos están firmemente establecidos.
A continuación, se explora cada pilar en mayor profundidad, abarcando las características básicas de cada pilar, destacando por qué es importante la transparencia dentro de este, qué progresos se han logrado hasta la fecha, las normas mundiales pertinentes que ya existen y las recomendaciones políticas para mejorar la transparencia de los ensayos clínicos.
REGISTRO DE LOS ENSAYOS
Todos los ensayos clínicos se deben inscribir en un registro aprobado por la OMS antes de enrolar al primer participante.
¿Qué es el registro de ensayos?
Los registros de los ensayos se crearon para proporcionar una visión rápida y completa de todos los ensayos clínicos realizados y en curso, y para reducir el sesgo al publicar y la distorsión de la evidencia. Actualmente existen 17 registros aprobados por la Organización Mundial de la Salud (OMS), todos ellos gestionados por entidades no comerciales y de libre acceso en línea. Los mayores registros son Clinicaltrials.gov (en EE UU), EudraCT (en la Unión Europea) y la Red de Registros Primarios de Japón, administrados por el sector público [14].
La inscripción de un ensayo consiste en diligenciar y cargar un breve formulario en un registro [15] que recoge información básica sobre el ensayo que se pretende realizar, incluyendo el fármaco o dispositivo médico que se quiere investigar, el número previsto de participantes y su problema médico, y los resultados específicos de salud que se evaluarán al final del ensayo para determinar los efectos, beneficios y riesgos del fármaco o dispositivo.
Una vez registrado el ensayo, los científicos de todo el mundo pueden ver lo que se ha investigado en el pasado y en qué están trabajando actualmente sus colegas. La adición permanente de un ensayo al mapa global de la investigación médica antes de que se inicie evita que los ensayos sin éxito desaparezcan por completo.
¿Por qué es importante?
Exigir que todos los ensayos se registren antes de comenzar garantiza que todos aquellos que se realicen con un fármaco o dispositivo sean y permanezcan visibles, independientemente de si los resultados son positivos o negativos, o de si se publican posteriormente o no. El registro de ensayos garantiza que ningún ensayo permanezca oculto y es indispensable para contrarrestar los efectos del sesgo de publicación (Zarin et al., 2007). Estos registros ya se utilizan ampliamente para obtener información sobre la investigación médica. En la actualidad, sólo el sitio Clinicaltrials.gov tiene aproximadamente 170 millones de visitas al mes (Zarin, 2017).
Además, dado que el registro prospectivo implica la especificación de las medidas de resultado (es decir, los criterios de éxito) de un ensayo por adelantado, reduce significativamente el potencial de distorsión de la evidencia posterior, es decir al presentar los resultados del ensayo (Dechartes et al., 2016).
Avances hasta la fecha
El registro anticipado de muchos tipos de ensayos clínicos ya es un requisito legal o reglamentario en muchas jurisdicciones, incluso en los EE UU (desde 2007; FDA, 2016) y en el Reino Unido (desde 2013; HRA, 2017). Sin embargo, no todos los ensayos clínicos están cubiertos en todas las jurisdicciones que cuentan con estos marcos regulatorios, y aún quedan algunos vacíos legales (Southworth, 2011).
Además, muchos actores importantes interesados en la investigación médica, incluyendo los financiadores de la investigación, las empresas farmacéuticas y las universidades, han adoptado políticas que promueven el registro de los ensayos [16]. En particular, una política adoptada por el Comité Internacional de Editores de Revistas Médicas dio lugar a un fuerte aumento en el número de ensayos registrados (Laine et al., 2007).
Como resultado de estos cambios, las tasas de registro de ensayos han aumentado en la última década, pero las investigaciones muestran que muchos ensayos siguen sin registrarse, incluso en jurisdicciones en las que el registro es obligatorio desde hace tiempo [17]. Alrededor de una tercera parte de los ensayos que sí se registran lo hacen de forma retroactiva, violando las normas éticas y los estándares de la Organización Mundial de la Salud, lo que deja la puerta abierta a la sustitución de los resultados y a otras formas de distorsión de la evidencia (Zarin et al., 2017). En otros casos, los datos proporcionados resultan de mala calidad (Zarin et al, 2011). Todo lo anterior indica que las leyes, las normas y los reglamentos existentes no se aplican adecuadamente.
|
Muchos ensayos siguen sin registrarse El registro prospectivo de los ensayos es una obligación ética universal para los investigadores médicos de todo el mundo desde 2008 (WMA, 2013). Sin embargo, un estudio de 2017 sobre 860 ensayos clínicos reveló que 556 ensayos no estaban registrados en absoluto, y otros 157 solo se habían registrado retroactivamente. Menos del 19% de los ensayos clínicos evaluados se habían registrado de manera prospectiva (Jones et al., 2017). |
|
En el Reino Unido, numerosos ensayos no se registran y no se imponen sanciones Todos los ensayos realizados en el Reino Unido tienen que obtener la aprobación de uno de los 68 comités regionales de ética de la investigación que tiene el país. Desde 2013, la aprobación ética se concede solo a los investigadores que se comprometen a registrar un ensayo. En 2015, la Autoridad de Investigación en Salud (Health Research Authority, HRA) del Reino Unido, que supervisa todos los comités de ética de la investigación, realizó una auditoría para descubrir cuántos ensayos no se habían registrado. Este organismo gubernamental descubrió que el 23% de los ensayos de fase 1, el 40% de los ensayos de dispositivos médicos y el 40% de “otros” ensayos no se habían registrado a tiempo. La HRA anunció que realizaría un seguimiento enviando un correo electrónico a los patrocinadores de los ensayos que seguían sin registrarse, pero asimismo señaló que “no existen sanciones de la HRA en este momento” (HRA, 2017). Posteriormente, un alto funcionario de la HRA explicó que el organismo carece de recursos y del marco jurídico necesarios para lograr eficazmente que se cumpla la normativa vigente (Kolstoe et al., 2017). |
Normas mundiales
Recomendaciones de política
Ya hay una serie de directrices internacionales sólidas, así como legislación y políticas aplicables en las jurisdicciones nacionales. Sin embargo, el principal obstáculo para lograr el registro universal proactivo y retroactivo de los ensayos es la falta de seguimiento y de sanciones eficaces cuando se incumplen estos requisitos. En muchas jurisdicciones, la aplicación efectiva de las leyes, normas y reglamentos existentes garantizaría que en el futuro todos los ensayos se registraran antes de comenzar.
Cuando no hay legislación o política, se puede utilizar como estándar de referencia a la Organización Mundial de la Salud para generar instrumentos sólidos.
Asimismo, la autoridad que los reguladores otorgan a los comités de ética, que son los que dan la aprobación para realizar los ensayos clínicos, pueden actuar como cuello de botella para garantizar el registro de los ensayos. Obligar a registrar el ensayo como requisito para la aprobación ética lograría que los ensayos se registraran antes de su inicio.
PUBLICACIÓN DE UN RESUMEN DE RESULTADOS
Se debe publicar un resumen de los resultados de todos los ensayos clínicos en los registros en los que se inscribieron originalmente durante los 12 meses siguientes a la conclusión del estudio [18].
¿Qué es un resumen de resultados?
Una vez finalizado el ensayo clínico, el resumen de sus resultados se publica en el registro (o registros) en el que se inscribió originalmente para poner a disposición del público la información básica sobre el desarrollo y los resultados del ensayo. El registro inicial de un ensayo recoge lo que los investigadores querían hacer y la metodología que proponían; posteriormente, el resumen de los resultados complementa esta información sintetizando las características y los hallazgos clave del ensayo en un breve cuadro [19]. El resumen de los resultados se puede publicar rápidamente y con un costo insignificante [20].
¿Por qué es importante?
El acceso al resumen de resultados permite que los científicos localicen, accedan y compartan rápida y sistemáticamente los nuevos descubrimientos. Además, los financiadores de la investigación pueden recurrir a los resúmenes de resultados para tomar mejores decisiones sobre la financiación de investigaciones adicionales sobre un nuevo fármaco o dispositivo. El resumen de resultados también reduce el potencial de sesgo al informar y la distorsión de la evidencia, ya que los resultados preespecificados del ensayo, los resultados reales y los hallazgos reportados en las publicaciones académicas se pueden comparar para verificar su consistencia (Rosati et al., 2016; Wieseler et al., 2012). Es importante destacar que el resumen de los resultados suele ofrecer una imagen más sólida y precisa de los resultados del ensayo, incluyendo los eventos adversos graves [21], que los artículos en las revistas (Riveros et al., 2013; Tang et al., 2015).
|
Los efectos adversos graves se documentan mejor en los resúmenes de los resultados Según la legislación estadounidense, se deben incluir todos los efectos adversos graves sufridos por los participantes en el ensayo (como la muerte, las afecciones que requieren hospitalización y los daños o discapacidades permanentes) en el resumen de resultados publicado en el registro Clinicaltrials.gov (FDA, 2016). Un estudio de 2015 descubrió que estos resúmenes de resultados proporcionaban una imagen más completa de los acontecimientos adversos graves que los trabajos publicados en las revistas académicas. Un equipo de investigación analizó 300 ensayos clínicos que habían informado sobre eventos adversos graves (EAG) en sus resúmenes de resultados y los comparó con los correspondientes artículos de revistas. Sólo 33 publicaciones en revistas (el 11%) ofrecían una descripción completa y coherente de todos los EAG sufridos por los participantes en el ensayo. Sin la disponibilidad del resumen de resultados, ninguno de los EAG registrados en el 41% de los ensayos se habría hecho público, ya que sus resultados no se habían publicado en medios académicos, o las publicaciones omitían mencionar los EAG. En el resto de los casos, los artículos de las revistas ofrecían una descripción imperfecta de los EAG observados. Además, el estudio descubrió que los resúmenes de resultados que se habían publicado informaban sobre los EAG mucho más rápidamente que los artículos de las revistas debido al tiempo que se tarda en procesar las publicaciones académicas. Los autores concluyeron que: “Para los responsables de la formulación de políticas, nuestros resultados promueven la ampliación de la publicación obligatoria de los resúmenes de resultados de los ensayos a todos los países… Es crucial consultar los resultados de seguridad que se publican en ClinicalTrials.gov […] para obtener más información sobre los perjuicios graves” (Tang et al., 2015). |
Un aspecto importante es que los investigadores pueden publicar los resúmenes de los resultados a los registros tan pronto como se hayan analizado los datos del ensayo, lo que acelera el ritmo del progreso científico. Tanto las normas mundiales como las leyes y reglamentos vigentes suelen exigir que los resúmenes de resultados se publiquen en un plazo máximo de 12 meses. En el caso de los ensayos pediátricos y las emergencias de salud pública, a veces se aplica un plazo más corto (UE, 2009; OMS, 2015). En cambio, conseguir que un artículo se publique en una revista académica puede llevar varios años (Tang et al., 2015).
|
Acelerar el ritmo de los descubrimientos médicos En 2016, dos empleados de una empresa farmacéutica publicaron un estudio que mostraba que 67 de los 69 ensayos clínicos concluidos exitosamente por la empresa farmacéutica en 2010 se habían presentado a revistas académicas para su publicación. Sin embargo, tres años tras la finalización de los ensayos, solo el 54 % de los estudios se habían publicado. La razón principal era la lentitud del proceso de publicación académica. Incluso en casos en los que la primera revista a la que se acudió aceptó el artículo, el promedio de tiempo hasta la publicación fue de 28 meses (Mooney y Fay, 2016). Por el contrario, los registros de ensayos clínicos suelen tardar sólo unas semanas en revisar y publicar el resumen de los resultados. Por tanto, la publicación de los resúmenes de los resultados permite a los científicos compartir los hallazgos de las investigaciones con mucha más rapidez. |
Publicar los resultados impide también que los descubrimientos médicos se pierdan para siempre en caso de que los resultados de un ensayo no lleguen a publicarse en una revista, por ejemplo, cuando los investigadores se trasladan a otra institución o se jubilan poco después de finalizar el ensayo. Un equipo de investigación dirigido por el director de Clinicaltrials.gov estimó recientemente que en la literatura se publican los resultados de solamente la mitad de los ensayos registrados (Zarin et al., 2017).
Avances hasta la fecha
La publicación oportuna de los resúmenes de resultados es una obligación ética y científica establecida por la OMS y por diversas normas internacionales. Además, en algunas jurisdicciones, las leyes, normas y reglamentos ya exigen la publicación oportuna de los resúmenes de resultados de algunos ensayos clínicos, aunque no de todos, en los registros de ensayos.
Por ejemplo, en EE UU, una ley de 2007 [22] exige a la “parte responsable”, por lo general a los patrocinadores de los ensayos [23], que publiquen el resumen de los resultados de ciertos ensayos clínicos en Clinicaltrials.gov [24], un registro de ensayos administrado públicamente, en un plazo de 12 meses tras la conclusión del ensayo. La ley establece que los que no cumplan la norma deberán pagar una multa de hasta US$10.000 por día hasta que se publiquen los resultados atrasados. Sin embargo, en la práctica esta ley nunca se ha aplicado. Varios años después de su aprobación, el estudio de una cohorte de ensayos sujetos a la notificación obligatoria reveló que el 78% no había cumplido con el requisito legal de publicar el resumen de resultados (Prayle, Hurley & Smyth, 2012) [25]. Hasta la fecha, tan solo los patrocinadores de ensayos comerciales han acumulado más de US$25.000 millones en multas por no publicar el resumen de los resultados, pero aún no se ha cobrado ninguna de ellas (Piller, 2015; Piller, 2016).
El índice global de publicación de los resúmenes de los resultados en Clinicaltrials.gov (es decir, incluyendo los ensayos no contemplados en la ley) es aún más bajo. Un estudio de 2013 sobre un conjunto de ensayos con medicamentos oncológicos descubrió que solo el 9% había publicado los resultados resumidos en el plazo de 12 meses; dos tercios de los ensayos aún no habían publicado los resultados tres años después de su conclusión (Nguyen et al., 2013). En octubre de 2016, ClinicalTrials.gov contaba con más de 227.000 registros, pero el 90% carecía de resúmenes de sus resultados (Anderson et al., 2015; Riveros et al., 2013; Zarin et al., 2017).
|
Se desconocen los resultados de 96 ensayos clínicos pediátricos En 2016, investigadores de la Facultad de Medicina de la Universidad de Harvard publicaron un estudio en el que se examinaban todos los ensayos pediátricos registrados en Clinicaltrials.gov entre 2008 y 2010. De los 455 ensayos concluidos, 136 no habían publicado sus resultados en revistas académicas. Gracias a los resúmenes de resultados publicados en ClinicalTrials.gov, los científicos tienen acceso a los resultados principales de 42 de esos ensayos, a pesar de no haberse publicado en la literatura académica. Sin embargo, los resultados de los otros 96 ensayos no publicados, en los que participaron decenas de miles de niños, siguen sin conocerse por completo y se pueden perder para siempre a menos que se publiquen los resúmenes de sus resultados (Pica y Bourgeois, 2016) [26]. |
Asimismo, un reglamento de la Unión Europea que entró en vigor en 2014 exige que los patrocinadores publiquen los resultados de ciertos ensayos [27] en un lapso máximo de 12 meses (seis meses para los ensayos pediátricos) (EMA, 2014). Sin embargo, aunque todos los ensayos inscritos en el registro administrado por la UE, EudraCT, están sujetos a esta ley, alrededor de un tercio de los ensayos que figuran en él en este momento carecen de resultados [28] . Esto sugiere claramente que las agencias nacionales [29] de los distintos países de la Unión Europea encargadas de hacer cumplir la normativa de la UE no la están aplicando eficazmente.
Normas globales
Recomendaciones de política
Aunque ya existen leyes, normas y reglamentos pertinentes en algunas jurisdicciones, no siempre se cumplen debido a la falta de supervisión y exigencia por parte de los organismos gubernamentales encargados de hacerlo [31]. Además, los marcos regulatorios existentes sólo contemplan algunos tipos de ensayos clínicos. Por ejemplo, según la normativa actual de la UE, no es obligatorio publicar los resultados de los ensayos con dispositivos médicos. Asimismo, en la actualidad algunos de los registros de ensayos no cuentan con una función que permita publicar los resúmenes de los resultados [32].
La ampliación del ámbito de aplicación de las leyes, normas y reglamentos existentes y su puesta en práctica de forma efectiva garantizarían que, en el futuro, los resúmenes de los resultados de todos los ensayos clínicos se publicaran en los registros en un plazo máximo de 12 meses tras la conclusión del ensayo, y que se publicaran retroactivamente los resúmenes de los resultados de todos los ensayos realizados desde la entrada en vigor de las leyes pertinentes. Para hacer cumplir la ley de manera efectiva, los organismos gubernamentales responsables [33] necesitarán tener un apoyo político fuerte y sostenido, facultades legales claras y apropiadas, una infraestructura técnica adecuada y recursos humanos suficientes (Kolstoe et al., 2017).
Obtener los resúmenes de los resultados de los ensayos más antiguos es igualmente importante, pero requerirá un acercamiento diferente. Muchos de los medicamentos que se utilizan hoy en día se desarrollaron en los años 90 o antes, por lo que los resultados de los ensayos clínicos más antiguos son muy relevantes para la práctica médica actual. El registro retroactivo de estos ensayos más antiguos y la publicación de los resúmenes de sus resultados mejoraría la prestación de los servicios médicos y la toma de decisiones por los organismos gubernamentales sobre la asignación de recursos, además de evitar que se pierdan para siempre las investigaciones médicas con un valor de miles de millones de dólares.
Se podría exigir a las empresas farmacéuticas que registraran y publicaran los resultados de los ensayos que han patrocinado en el pasado, como condición para seguir accediendo al mercado [34]. Los resultados de muchos ensayos antiguos, patrocinados por instituciones no comerciales, como las académicas, se podrían obtener mediante una combinación de incentivos financieros y asistencia técnica.
Las inversiones necesarias para garantizar que se comuniquen los resultados de los ensayos clínicos pasados y presentes son minúsculas en comparación con los costos de llevar a cabo la investigación, y resultan insignificantes en comparación con los beneficios científicos y de salud pública y con una asignación de más amplia de recursos si estos datos están disponibles [35].
INFORMES COMPLETOS DE LOS ENSAYOS
Toda la información pertinente para interpretar los resultados de un ensayo se debe divulgar de forma proactiva y se debe poner a disposición de la comunidad científica. Esta información incluye el protocolo original del ensayo, un plan de análisis estadístico preespecificado, formularios de informes de casos e informes de estudios clínicos (IEC o CSR).
¿En qué consisten los informes completos sobre los ensayos?
Los informes completos sobre los ensayos contienen la información que los investigadores necesitan para comprender plenamente el desarrollo y los resultados de un ensayo [36]. El componente principal de los informes completos de ensayos en entornos comerciales son los informes de estudios clínicos (IEC o CSR), documentos eminentemente técnicos que suelen tener más de 1.000 páginas y ofrecen una imagen muy detallada del diseño, el desarrollo, el análisis y los resultados de un ensayo, incluyendo los detalles sobre los efectos secundarios negativos sufridos por cada paciente. Por lo tanto, estos informes ofrecen una visión de la metodología y los hallazgos de un ensayo mucho más profunda que los resúmenes de resultados o los artículos publicados en revistas académicas (Doshi et al., 2012).
Cuando las empresas farmacéuticas solicitan una licencia para comercializar un nuevo medicamento, deben presentar los informes de los estudios clínicos (IEC) pertinentes a los organismos reguladores, como la FDA o la EMA para su revisión. Para facilitar el proceso, la Conferencia Internacional de Armonización establece un formato y una estructura estandarizados a nivel mundial que deben observar todos los IEC (ICH, 2015). En cambio, los informes de ensayos completos que se elaboran en entornos no comerciales pueden no ajustarse a un formato estandarizado.
¿Por qué es importante?
La gran cantidad de detalles que contienen los informes completos de los ensayos permite a los científicos “hacer un análisis más detallado” y comprender exactamente cómo se llevó a cabo un ensayo clínico, calibrar la exactitud, la fiabilidad y la validez de los hallazgos del ensayo, y obtener mucha más información sobre los beneficios y los efectos secundarios de los medicamentos que la que se puede obtener en los resúmenes de los resultados o en los artículos de revistas (Jefferson et al., 2014).
Sin acceso a los IEC completos, incluyendo todos los apéndices, la comunidad científica no puede verificar plenamente la exactitud, la fiabilidad y la validez de los resultados de los ensayos, ni detectar muchos tipos de omisiones, errores, interpretaciones equivocadas, distorsión de la evidencia y tergiversaciones en otros tipos de información sobre los ensayos (Doshi y Jefferson, 2013) [37].
|
Los efectos secundarios mortales suelen permanecer ocultos En 2016, un metaanálisis de 28 estudios distintos sobre el subregistro de daños descubrió que más del 60 % de los efectos secundarios negativos que se detectaron durante los ensayos clínicos y se registraron en documentos no publicados no se recogen en las revistas académicas. Los 28 estudios sobre el tema, sin excepción, habían identificado un mayor número de eventos adversos (o de todos los adversos graves) en las versiones no publicadas. En un ejemplo, se registraron 198 muertes en los ensayos clínicos con cuatro fármacos nuevos, pero en los documentos publicados posteriormente sólo se informó exhaustivamente sobre 29 muertes. En otro ejemplo, un informe no publicado documentó 15 suicidios, pero en los documentos publicados sólo se dieron a conocer siete. El meta-análisis concluyó que “la extensión de los datos ‘ocultos’ o ‘perdidos’ impide que los investigadores, los médicos clínicos y los pacientes tengan una comprensión completa del daño, y esto puede llevar a hacer juicios incompletos o erróneos” (Wieseler et al., 2010). |
Sobre todo, los organismos de evaluación de tecnologías aplicadas a la salud pública, responsables de la evaluación de las propiedades, los efectos y los impactos de las tecnologías en salud, necesitan ser capaces de revisar los IEC para mejorar su toma de decisiones sobre la eficacia clínica, la seguridad y la rentabilidad de las diferentes opciones de tratamiento (Wieseler et al., 2010; Wieseler et al., 2012).
|
Los IEC son “esenciales” para la toma de decisiones informadas en materia de política en salud La agencia alemana de evaluación de tecnologías aplicadas a la salud, IQWiG, habitualmente solicita los IEC a los fabricantes para hacer sus evaluaciones de medicamentos, las cuales informan la toma de decisiones sobre políticas en el sistema de salud alemán. En 2013, un equipo de IQWiG revisó 101 ensayos clínicos cuyos IEC completos habían sido suministrados voluntariamente por las empresas farmacéuticas. Descubrieron que los IEC proporcionaban más del doble de la información sobre los resultados pertinentes para los pacientes que todas las demás fuentes disponibles públicamente juntas. Los científicos del IQWiG concluyeron que los IEC son “fuentes esenciales para fundamentar comparaciones indirectas significativas [entre diferentes medicamentos]”. Señalando que actualmente las empresas farmacéuticas no están obligadas a proporcionar los IEC a las agencias de evaluación de tecnologías médicas o a otras terceras partes, recomendaron que “los IEC se pusieran a disposición del público” (Wieseler et al., 2013). |
Además, los investigadores independientes necesitan que los IEC verifiquen de manera independiente el resumen de la evidencia de los ensayos que se presentan y cómo se generan, y que reevalúen las conclusiones a las que llegan las empresas farmacéuticas y las agencias reguladoras.
|
Cómo la transparencia en los IEC mejoró la toma de decisiones en materia de reglamentación En 2007, investigadores independientes analizaron los IEC de los ensayos clínicos de un medicamento para la diabetes muy utilizado y concluyeron que el fármaco se asociaba a un mayor riesgo de accidentes cerebrovasculares, infartos de miocardio y muertes por causas cardíacas. La empresa que comercializaba los medicamentos había puesto los IEC a disposición de investigadores externos en el transcurso de un litigio. Los reguladores, tanto en EE UU como en Europa, reaccionaron rápidamente modificando sus evaluaciones previas sobre la seguridad del fármaco; al menos un regulador recomendó retirarlo del mercado por completo (CRIT, 2017). |
Poner a disposición del público los informes completos de los ensayos – incluyendo, entre otros, los IEC – reduce la posibilidad de errores, interpretaciones erróneas, sesgos, distorsión de la evidencia, corrupción o fraude en otras formas de información sobre los ensayos (Doshi y Jefferson, 2013). Por otra parte, el acceso a los informes completos de los ensayos puede ayudar a los expertos independientes a señalar los beneficios o perjuicios que el equipo que realizó originalmente el ensayo puede haber pasado por alto o haber presentado de forma incompleta, mejorando así la seguridad de los pacientes [38] y acelerando el descubrimiento de nuevos tratamientos y curas (Association of Medical Research Charities, 2016).
|
El uso de los IEC para sacar a la luz los daños no reconocidos de los medicamentos Un equipo independiente de investigadores analizó siete informes de la EMA que no se habían divulgado antes y descubrió datos sobre efectos adversos que no se habían recopilado ni comunicado en su totalidad. Todos los IEC estaban relacionados con el orlistat, un fármaco diseñado para tratar la obesidad que se puede adquirir sin receta en EE UU y en muchos países europeos. En su estudio de 2016, los investigadores concluyeron que los daños habían sido “subestimados sistemáticamente ” no solo en los artículos académicos, sino también en los resultados resumidos presentados a la EMA en el proceso de aprobación del medicamento. Dado que los IEC se presentaron mucho antes de que entrara en vigor la política de divulgación proactiva de la EMA, los investigadores tuvieron que recurrir a solicitudes de libertad de información para acceder a ellos, procedimiento que duró casi cuatro años. Tras revisar los datos, el autor principal concluyó que “es muy poco probable que la EMA descubriera la diferencia en la duración de los eventos adversos en el grupo de orlistat y en el del placebo. La EMA se basa en el análisis desarrollado por el patrocinador y normalmente no realiza su propio análisis estadístico” (Schroll, Penninga y Gøtzsche, 2016). |
Avances hasta la fecha
Hace tiempo que las empresas farmacéuticas están obligadas a compartir los IEC pertinentes con los reguladores cuando solicitan las licencias para comercializar medicamentos nuevos. Sin embargo, en la mayoría de los casos, sólo la empresa farmacéutica que comercializa un medicamento y los funcionarios que trabajan para las agencias reguladoras pueden acceder a los IEC. En cambio, los científicos que trabajan para otros organismos gubernamentales, como las agencias de tecnología médica y de salud pública, a menudo no pueden acceder a estos importantes documentos, como tampoco lo hacen los investigadores independientes (Gøtzsche y Jørgensen, 2011; Wieseler et al., 2013).
En 2016, la EMA abrió una nueva brecha al publicar de forma proactiva algunos IEC. Lamentablemente, la nueva política solo cubre algunos informes recibidos por el organismo regulador desde 2015 [39]. Los IEC más antiguos, en particular, quedan fuera de la política y, por lo tanto, permanecen en los archivos de la agencia, por lo que la gran mayoría de ellos, en poder de la EMA, siguen siendo inaccesibles. Esto deja abiertos interrogantes sobre muchos medicamentos que se utilizan con frecuencia (AllTrials, 2016). Por otra parte, la EMA no pone a disposición del público los IEC de manera que puedan ser descargados y compartidos libremente por cualquier persona. En la actualidad, estos informes solo se pueden ver en la pantalla y solo por los científicos a los que se les ha concedido acceso previa solicitud, lo que limita su utilidad. A pesar de estas salvedades, la medida de la EMA constituye un paso audaz y significativo en la dirección correcta.
Algunas empresas farmacéuticas han llevado a la EMA a los tribunales para impedir la publicación de información adicional (Wieseler et al., 2013). La industria farmacéutica suele argumentar que la publicación de los IEC podría vulnerar la confidencialidad de los pacientes o revelar secretos comerciales, pero la EMA consideró que estas cuestiones se podían solucionarse tachando una cantidad número muy limitada de información [40].
En EE UU, la FDA no tiene una política de transparencia comparable, lo que impide a terceros acceder a los IEC presentados al organismo regulador estadounidense, excepto a través de solicitudes basadas en la ley de libertad de información.
Los informes completos de ensayos que no se recopilan para el uso de los reguladores, entre ellos muchos producidos por investigadores que trabajan en universidades e instituciones de investigación sin ánimo de lucro, tampoco suelen ponerse a disposición del público. Por lo general, los científicos que se desempeñan en estos contextos carecen de incentivos para publicar los informes completos de los ensayos, y no hay mecanismos debidamente establecidos para permitir el acceso del público. Por ejemplo, los registros de ensayos más utilizados carecen de una función específica que permita cargar los informes completos de los ensayos.
Normas mundiales
Recomendaciones de política
Los organismos reguladores de todo el mundo deberían seguir y ampliar el ejemplo positivo establecido recientemente por la EMA. En concreto, deberían poner a disposición del público todos los IEC (incluyendo todos los apéndices) que actualmente se encuentran en sus archivos, después de realizar algunas tachaduras para salvaguardar la confidencialidad comercial y la de los pacientes. En el futuro, los reguladores deberían hacer públicos los nuevos IEC en el momento de la aprobación reglamentaria, o en un plazo máximo de 24 meses desde la recepción del IEC si no se ha aprobado.
Los organismos reguladores ya tienen archivados estos y otros documentos, por lo que publicarlos en línea no plantea problemas logísticos y puede hacerse con un costo mínimo (Turner, 2007) [43]. Desde el punto de vista legal, estas disposiciones en materia de transparencia se podrían aplicar condicionando la continuidad de cualquier medicamento al mercado a la publicación de todos los IEC pertinentes [44].
Dada la resistencia activa de algunos actores de la industria a una mayor transparencia en este ámbito, las agencias reguladoras, que por lo general apoyan las medidas de transparencia, necesitarán un apoyo político firme y sostenido a lo largo de este proceso (EMA, 2016).
LA PUBLICACIÓN ACADÉMICA
Los resultados de todos los ensayos clínicos se deben publicar en una revista académica o se deben poner a disposición del público en registros o en bases de datos de ensayos adecuados (preferiblemente en ambos).
¿En qué consiste la publicación académica?
Los investigadores suelen compartir los hallazgos de un ensayo clínico a través de la publicación de un artículo que resume el diseño y los resultados del ensayo en una revista científica. Si la revista está interesada en publicar el artículo, lo envía a otros científicos y expertos en la materia para que lo revisen y pide al autor que haga las modificaciones necesarias al artículo en función de los comentarios recibidos y lo vuelva a presentar. Este proceso puede llevar mucho tiempo, pero muchos científicos lo consideran un mecanismo esencial para garantizar la calidad.
¿Por qué es importante?
Las revistas científicas son la principal plataforma de comunicación para muchos investigadores de todo el mundo y a menudo son el primer recurso para los médicos que buscan información sobre la mejor manera de tratar a sus pacientes. De ahí que, para garantizar que los descubrimientos logrados por los ensayos clínicos se compartan ampliamente y mejoren la práctica médica, los hallazgos de todos los ensayos clínicos se deberían publicar en una revista académica o ponerse a disposición del público en registros o bases adecuadas de datos de ensayos (preferiblemente en ambos). Sin embargo, muchos ensayos no publican sus resultados. En un estudio de 2012 de una serie de ensayos financiados con fondos públicos en EE UU se descubrió que alrededor de 60.000 personas habían participado en ensayos que nunca se publicaron (Asiimwe y Dickson, 2016; Hwang, et al., 2016; Pica y Bourgeois, 2016; Ross et al., 2012).
Muchos otros ensayos se publican en revistas sólo para suscriptores o de pago por artículo, lo que limita el acceso a los resultados de una investigación importante y a menudo financiada con fondos públicos. En algunos casos, un artículo puede tardar varios años en publicarse en una revista. Además, los artículos académicos suelen omitir datos importantes (Duff et al., 2010). Y, lamentablemente, múltiples estudios muestran que la literatura académica pinta una imagen sistemáticamente sesgada y frecuentemente inexacta de la seguridad y la eficacia de los medicamentos (Golder et al., 2016).
Avances hasta la fecha
La comunidad de investigadores médicos ha ideado múltiples formas de superar la falta de informes, el sesgo y la mala conducta de los investigadores en las publicaciones académicas, pero su aplicación en la práctica ha sido difícil debido al gran número de personas e instituciones involucradas, al panorama fragmentado de las publicaciones y a incentivos perversos. En la actualidad existen revistas comprometidas con la publicación de ensayos con resultados nulos o negativos, revistas de prepublicación rápida, revistas que ofrecen acceso abierto a los artículos de forma gratuita y revistas que aceptan “informes registrados”, pero los incentivos profesionales que ofrece actualmente el mundo académico suelen desanimar a los científicos a publicar en ellas (Goldacre et al., 2016). Por otra parte, los científicos que no publican sus resultados no se enfrentan a ninguna sanción, y es poco probable que se descubra a los que distorsionan la evidencia y aún menos probable que enfrenten a sanciones reales; incluso la falsificación fraudulenta de datos suele quedar impune (Doshi, 2015).
|
La distorsión de la evidencia: el cambio de las medidas de resultado, ¿la excepción o la norma? El “cambio de las medidas de resultado” es una forma de distorsionar la evidencia que consiste en modificar los objetivos de un ensayo una vez concluido. Para usar una analogía, los investigadores lanzan sus flechas primero y luego dibujan un blanco alrededor de donde han caído las flechas, haciendo que su tiro con arco parezca mucho más impactante de lo que realmente es. En esencia, informar sobre los éxitos en el tratamiento de los pacientes basándose en datos de ensayos que han cambiado las medidas de resultado hace que los medicamentos y dispositivos parezcan mucho más eficaces de lo que realmente son. En 2015-2016, un equipo de investigadores de la Universidad de Oxford revisó los artículos publicados en las cinco principales revistas médicas del mundo para averiguar qué tan común es el cambio en las medidas de resultados. Muchos médicos confían en estas revistas para obtener orientación sobre cómo tratar a sus pacientes. El equipo de Oxford descubrió que las publicaciones de 58 de 67 ensayos habían modificado los resultados. En total, 354 resultados preespecificados no se habían publicado, mientras que se habían añadido discretamente 357 resultados nuevos. Sólo nueve de los 67 ensayos se habían publicado correctamente. Los investigadores de Oxford descubrieron múltiples casos de distorsión de la evidencia al comparar los resultados publicados en las revistas médicas con aquellos previamente consignados en los registros de ensayos clínicos. Si estos ensayos no se hubieran registrado, habría sido imposible detectar la información engañosa sobre algunos medicamentos y dispositivos (Goldacre, Drysdale y Powell-Smith, 2016). |
Normas globales
Recomendaciones de política
Los gobiernos nacionales tienen la capacidad de configurar el panorama de la investigación médica. Al mismo tiempo, la mayoría se ha mostrado reticente a la hora de tomar medidas que se puedan percibir como una intromisión en la libertad académica [46]. No obstante, los gobiernos podrían reducir los sesgos y la distorsión de la evidencia en la literatura médica sin intervenir directamente en el ámbito académico.
En algunas jurisdicciones, los organismos públicos que supervisan los comités de ética en la investigación están bien posicionados para monitorear la falta de publicación de los resultados de los ensayos y algunas formas de distorsión de la evidencia en las publicaciones utilizando los registros que ya conservan en su archivo (Chan et al., 2017) [47].
Además, dado que muchos ensayos clínicos se financian con el dinero de los contribuyentes, los responsables de las decisiones políticas podrían dar instrucciones a los organismos públicos de financiación para que supervisen activamente las publicaciones de los beneficiarios y sancionen a las instituciones cuyas publicaciones no se ajusten a las mejores prácticas. Algunos financiadores ya han empezado a aplicar este tipo de sistemas de control [48].
COMPARTIR LOS DATOS DE LOS PARTICIPANTES INDIVIDUALES
Se deben establecer estructuras, normas, políticas y legislación que permitan compartir los datos de los participantes de forma eficaz y controlada.
¿En qué consiste el compartir los datos de los participantes individuales?
Los datos de participantes individuales (DPI), o datos de pacientes individuales, son los datos recaudados sobre cada participante en el transcurso de un ensayo clínico. Compartir los DPI significa que, una vez concluido el ensayo, los investigadores ponen a disposición de otros científicos estos datos de carácter individual.
¿Por qué es importante?
Compartir los datos de los ensayos clínicos tiene un gran potencial para acelerar el progreso científico (Zarin y Tse, 2016). Al agregar los datos de múltiples ensayos, los investigadores pueden generar mejor evidencia sobre la seguridad y la eficacia de los medicamentos, dispositivos y tratamientos (Debray et al., 2016). Asimismo, los científicos pueden desglosar los datos de múltiples ensayos y recombinarlos, por ejemplo, para explorar las variaciones en los efectos del tratamiento en diferentes subgrupos de la población. La puesta en común de datos también puede ofrecer oportunidades adicionales para la investigación exploratoria, que podría conducir a nuevos descubrimientos científicos, a tratamientos más eficaces o a usos alternativos de los tratamientos existentes (Tierney et al., 2015).
Además, los investigadores independientes pueden utilizar los DPI para llevar a cabo nuevos análisis de los datos generados en un ensayo. La repetición del análisis de los datos de los pacientes por expertos independientes reduce la posibilidad de errores, interpretaciones erróneas, distorsión de la evidencia y el fraude que se producen en otras formas de presentación de informes de los ensayos (Ross, 2016).
|
Cómo el intercambio de DPI contribuyó a que más niños pequeños sobrevivieran a la malaria Se calcula que la malaria mata a medio millón de personas cada año, muchas de ellas niños. Sin embargo, los médicos llevan mucho tiempo sin saber cuál es la dosis óptima de un tratamiento comúnmente utilizado en niños pequeños. Un análisis de 2013 que agrupaba los DPI de 26 ensayos clínicos diferentes demostró que los niños de 1 a 5 años tenían más probabilidades de recuperarse si se les administraban dosis más altas del tratamiento. Este descubrimiento habría sido imposible de hacer analizando un solo estudio (CRIT, 2017) |
Avances hasta la fecha
Si bien este pilar de la transparencia en los ensayos clínicos se considera un asunto delicado, ya que requiere que se preste atención especial a la protección robusta de los datos, numerosos financiadores de investigación y empresas farmacéuticas han puesto en marcha políticas de intercambio de DPI, y muchos investigadores que realizan ensayos clínicos han manifestado su compromiso con compartir voluntariamente los DPI (Bergeris et al., 2017; Smith et al., 2017; Storm, 2014; Wellcome Trust, 2015). La FDA de EE UU ya exige a las empresas farmacéuticas que presenten los DPI, que luego la agencia analiza para evaluar mejor los beneficios y daños de los medicamentos (CRIT, 2017).
Sin embargo, los esfuerzos concertados para que compartir los DPI sea la norma son relativamente recientes, y siguen existiendo considerables obstáculos para compartir los DPI. Entre los obstáculos prácticos se encuentran el importante esfuerzo que hay que hacer para convertir los datos brutos en formatos que otros investigadores puedan utilizar, la actual ausencia de normas universalmente acordadas, los retos relacionados con la salvaguarda de la confidencialidad de los pacientes, las cuestiones legales y normativas y los retos metodológicos.
Debido a estas consideraciones, en 2017, el Comité Internacional de Editores de Revistas Médicas (ICMJE por sus siglas en inglés) descartó una propuesta para obligar a compartir los DPI a corto plazo, al tiempo que reafirmó su esperanza y convicción de que en el futuro compartir los DPI se convertiría en la norma (Taichman et al., 2016).
Mientras tanto, los defensores de la transparencia han señalado que las empresas tienden a ofrecer únicamente un acceso controlado a los solicitantes aprobados, en lugar de compartir los DPI como información de acceso abierto que se puede descargar y compartir libremente, y que la industria sigue considerando a los DPI como propiedad de los patrocinadores de los ensayos y no como parte de un patrimonio científico global.
Normas globales
La OMS apoya el desarrollo de estructuras, normas y estándares que permitan compartir los DPI de forma eficaz. Una consulta de la OMS realizada en septiembre de 2015 afirmó que el intercambio oportuno y transparente de datos y resultados durante las emergencias de salud pública se debe convertir en una práctica habitual (Modjarrad et al., 2016).
Recomendaciones de política
Existe un amplio consenso en la comunidad de la investigación médica en que, si se hace bien, el compartir los DPI puede contribuir significativamente al progreso médico. Este campo está evolucionando rápidamente, y en la actualidad se está llevando a cabo una serie de iniciativas prometedoras que los responsables políticos deberían fomentar y apoyar. Para garantizar que estos esfuerzos desarrollen todo su potencial, hay que adoptar legislación que garantice una sólida protección de los datos de los ensayos clínicos.
Por último, es importante destacar que el inmenso potencial de compartir los DPI de forma eficaz solo se puede aprovechar plenamente si los demás pilares de la transparencia en los ensayos clínicos están firmemente establecidos (Hoffmann et al., 2017; Zarin y Tse, 2016).
PRINCIPIOS Y MEDIDAS PRÁCTICAS PARA LOS RESPONSABLES DE LA FORMULACIÓN DE POLÍTICAS
La opacidad es muy cara
La opacidad en la investigación clínica ya ha costado innumerables vidas humanas y ha supuesto un importante despilfarro de fondos de salud pública. Como ha señalado una comisión parlamentaria del Reino Unido:
“Los fabricantes ocultan a los médicos e investigadores información importante sobre los ensayos clínicos de manera rutinaria y legal. Este prolongado incumplimiento normativo y cultural repercute en toda la medicina y socava la capacidad de los médicos, los investigadores y los pacientes para tomar decisiones informadas sobre qué tratamiento es el mejor.” Comité de Cuentas Públicas, 2013
Las medidas a favor de la transparencia son factibles y muy rentables
Desde el punto de vista positivo, la transparencia de los ensayos clínicos se puede reforzar significativamente utilizando los sistemas, procesos y herramientas existentes. En muchas jurisdicciones, ya existen leyes, normas y reglamentos apropiados, y los organismos públicos encargados de su aplicación suelen apoyar firmemente las medidas de transparencia. Pueden lograrse muchos avances significativos en materia de transparencia aplicando los marcos legales existentes, solo con medidas administrativas (US FDA, 2017). En muchos casos, los costos se podrían recuperar por completo mediante la imposición de multas por infracciones [49]. En otros casos, hacer que los ensayos clínicos sean más transparentes sería poco costoso como altamente rentable, como señaló recientemente una coalición de importantes financiadores de la investigación médica convocada por la OMS:
“Habrá costos modestos asociados a la divulgación pública de los resultados de los ensayos clínicos. Los costos de la difusión de los resultados son un componente menor de los costos generales de la investigación, y la divulgación de los resultados es un elemento esencial del trabajo de investigación. La asignación de recursos, los beneficios científicos y de salud pública, y la necesidad de cumplir con los imperativos éticos superan con creces los costos”. OMS, 2017 [50]
Un imperativo para la acción política
Los responsables de la toma de decisiones políticas en todo el mundo deben asumir la responsabilidad de resolver este acuciante problema de salud pública; es un asunto que repercute en la vida de todas las personas alrededor del mundo. En 2016, Naciones Unidas exigió:
“Los gobiernos deben disponer que los datos no identificables de todos los ensayos clínicos concluidos y suspendidos se pongan a disposición del público en un registro abierto de fácil consulta […] Para facilitar la colaboración abierta, la reconstrucción y una nueva investigación de los fracasos, los gobiernos deben exigir que los diseños y protocolos de los estudios, las bases de datos, los resultados de los ensayos y los datos de los pacientes protegidos por el anonimato estén a disposición del público de forma oportuna y accesible [… …] Para que el público obtenga todos los beneficios de la inversión pública en investigación, los organismos de financiación pública deben garantizar que, cuando sea factible, los datos, los resultados y los conocimientos generados por dicha inversión pública se pongan a disposición de todos […] El aumento de la transparencia de la información sobre los ensayos clínicos contribuye en gran medida a mejorar los resultados de la salud pública”. Panel de alto nivel de la ONU, 2016 [51]
Tres medidas para que los ensayos clínicos sean más transparentes
Los responsables de la toma de decisiones deberían adoptar las tres medidas siguientes para incrementar la transparencia de los ensayos clínicos y lograr que el sector responda mejor ante los ciudadanos, los pacientes, los contribuyentes y los inversionistas:
Primera medida: Garantizar la transparencia de los ensayos clínicos financiados con fondos públicos. Como primera medida, los responsables de la toma de decisiones políticas deberían exigir a todos los organismos públicos que financian investigación dentro de su jurisdicción que adopten y amplíen las disposiciones de la reciente “Declaración conjunta” de los financiadores de la investigación, negociada por la OMS, y que garanticen su plena aplicación. Para contribuir a garantizar que la financiación pública de la investigación médica beneficie efectivamente a los ciudadanos, los financiadores gubernamentales deberían destinar el dinero de los contribuyentes únicamente a instituciones e individuos que cumplan de forma verificable con las mejores prácticas de investigación clínica. Este sencillo primer paso supondría un importante aumento de la transparencia con costos mínimos.
Segunda medida: aplicar las normas vigentes. En segundo lugar, los responsables de la toma de decisiones deben dotar a los organismos gubernamentales de los recursos, las competencias y el apoyo político que necesitan para hacer cumplir las leyes, normas y reglamentos existentes, que en la actualidad no se suelen aplicar de forma coherente. Los responsables políticos deben apoyar a los entes gubernamentales en la creación de mecanismos eficaces de control y de imposición de sanciones para que el sector rinda cuentas. Un modelo de control prometedor consiste en utilizar los registros existentes de los comités de ética de la investigación para supervisar el registro, la divulgación de los resúmenes de resultados y la publicación académica de todos los ensayos realizados en una determinada jurisdicción [52].
Tercera medida: reforzar los marcos jurídicos y regulatorios. En tercer lugar, los responsables de la toma de decisiones deben adaptar las leyes, normas y reglamentos existentes [53] a las normas de mejores prácticas mundiales y garantizar que abarquen todos los ensayos clínicos, pasados y presentes, según la definición de la OMS. Por ejemplo, en la Unión Europea, las directrices actuales sobre la comunicación de resultados se deberían ampliar más allá de su actual concentración en determinados ensayos de medicamentos, y las políticas de divulgación de los informes de estudios clínicos se deberían ampliar para incluir los ensayos más antiguos. Para garantizar la transparencia de los ensayos clínicos, los marcos jurídicos y normativos deberían incorporar los cinco elementos siguientes:
Registro del ensayo: todos los ensayos clínicos deben inscribirse en un registro aprobado por la OMS antes de inscribir al primer participante.
Publicación de resúmenes de resultados: los resúmenes de resultados de todos los ensayos clínicos se deben publicar en los registros en los que se inscribieron originalmente en un plazo de 12 meses tras la conclusión del estudio.
Informes completos de los ensayos: se debe divulgar toda la información pertinente para interpretar los resultados de un ensayo de forma proactiva y se debe poner a disposición de la comunidad científica. Esta información debe incluir el protocolo original del ensayo, un plan de análisis estadístico preespecificado, los formularios de informe de casos y los informes del estudio clínico.
Publicación académica: Los resultados de todos los ensayos clínicos se deben publicar en una revista académica o ponerse a disposición del público en los registros o bases de datos de ensayos apropiados (preferiblemente en ambos).
Compartir los datos de los participantes individuales: Hay que establecer estructuras, normas, políticas y legislación que permitan compartir los datos de los participantes individuales de forma eficaz y controlada.
Notas
Referencias
Aizaid, A. Chasing a missing clinical trial result – a doctor’s odyssey, AllTrials, 2016. http://www.alltrials.net/news/ diabetes-alzheimer-link-clinical-trials/ [Accessed 10 November 2017].
AllTrials. Pharma company investors call for clinical trials transparency, AllTrials, 2015. http://www.alltrials.net/news/ pharma-company-investors-call-for-clinical-trials-transparency/ [Accessed 08 November 2017].
AllTrials, “European Medicines Agency today releases first Clinical Study Reports”, AllTrials, 2016. http://www. alltrials.net/news/european-medicines-agency-csr-transparency-policy-0070/ [Accessed 20 September 2017].
Anderson, L. M. et al. Compliance with Results Reporting at ClinicalTrials.gov, New England Journal of Medicine, 2015; 372: 1031-1039.
Angell, M. The Truth about the Drug Companies. How they deceive us and what to do about it, New York: Random House, 2004.
Asiimwe, I.G. & Rumona, D. Publication proportions for registered breast cancer trials: before and following the introduction of the ClinicalTrials.gov results database, Research Integrity and Peer Review, 2016; 1:10.
Baronikova, S. et al. Disclosure of results of clinical trials sponsored by pharmaceutical companies. Presented at the Peer Review Congress (10–12 September 2017, Chicago, USA). Available at: http://peerreviewcongress.org/ prc17-0354 and http://www.shirecongressposters.com/717649 [Accessed 11 September 2017]
Bass, A. Side Effects: A Prosecutor, a Whistleblower, and a Bestselling Antidepressant on Trial, New York: Algonquin, 2008. ISBN: 9781565125537
Begum, R. & Kolstoe, S. Can UK NHS research ethics committees effectively monitor publication and outcome reporting bias? BMC Medical Ethics, 2015. https://bmcmedethics.biomedcentral.com/articles/10.1186/s12910-015- 0042-8 [Accessed 20 September 2017]
Bekelman, J.E., Li, Y. & Gross, C.P. Scope and impact of financial conflicts of interest in biomedical research: a systematic review. JAMA, 2003; 289(4): 454-465.
Bergeris, A., Tse, T. & Zain, D. Statements About Intent to Share Individual Participant Data at ClinicalTrials.gov, International Congress on Peer Review and Scientific Publication, 2017. http://peerreviewcongress.org/prc17-0261 [Accessed 10 November 2017]
Bruckner, T. How we dragged orlistat’s hidden “understated harms” into the light: An interview with Jeppe Schroll, AllTrials, 2016. http://www.alltrials.net/news/orlistat-alli-jeppe-schroll-ema-foi-clinical-trials/ [Accessed 09 November 2017]
Bruckner, T. & Ellis, B. “Clinical Trial Transparency: A Key to Better and Safer Medicines”, TranspariMED, 2017; https://www.transparimed.org/single-post/2017/04/28/clinical-evidence-harms-costs [Accessed 8 November 2017]
Carpenter, D. Reputation and Power: Organizational Image and Pharmaceutical Regulation at the FDA, New Jersey: Princeton University Press, 2010.
Center for Open Science. Registered Reports: Peer review before results are known to align scientific values and practices, Center for Open Science. https://cos.io/rr/ [Accessed 26 September 2017]
Chalmers, I. et al. Testing Treatments, NIHR, 2017. http://www.testingtreatments.org/ [Accessed 30 November 2017]
Chan, A-W, et al. 2017. Association of Trial Registration With Reporting of Primary Outcomes in Protocols and Publications, JAMA Research Letter, 11 September 2017. http://jamanetwork.com/journals/jama/fullarticle/2653434 [Accessed 30 November 2017]
ClinicalTrails.gov & U.S. National Library of Medicine. FDAAA 801 Requirements, 2016. https://clinicaltrials.gov/ct2/manage-recs/fdaaa [Accessed 20 September 2017].
CRIT. Promoting Transparency in Clinical Research: Why and How? Yale University Press, 2017. https://law.yale.edu/system/files/area/center/crit/document/crit_white_paper_november_2017_best_.pdf [Accessed 08 November 2017]
Collier, R. Scientific misconduct or criminal offence? Canadian Medical Association Journal, 2015, 187(17): 1273–1274.
Crowther, C. et al. Assessing the neuroprotective benefits for babies of Antenatal Magnesium Sulphate: An individual participant data meta-analysis. PLoS Medicine, 2017, 14(10).
Davis, C. et al. Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: retrospective cohort study of drug approvals 2009-13, BMJ, 2017; 359:j4530.
Debray, T. et al. An overview of methods for network meta-analysis using individual participant data: when do benefits arise? Statistical Methods in Medical Research, 2016.
Dechartres, A. et al. Association between trial registration and treatment effect estimates: a meta- epidemiological study, BMC Medicine 2016; 14(1): 100.
Doshi, P., Jefferson T. & Del Mar C. The Imperative to Share Clinical Study Reports: Recommendations from the Tamiflu Experience, PLoS Medicine, 2012 9(4): e1001201.
Doshi, P., Jones, M. & Jefferson, T. Rethinking credible evidence synthesis, BMJ, 2012; 344:d7898
Doshi, P. & Jefferson, T. Clinical study reports of randomised controlled trials: an exploratory review of previously confidential industry reports, BMJ Open, 2013; 3 e002496.
Doshi, P. From promises to policies: Is big pharma delivering on transparency? BMJ, 2014; 348: g1615.
Doshi, P. No correction, no retraction, no apology, no comment: paroxetine trial reanalysis raises questions about institutional responsibility, BMJ, 2015; 351:h4629.
Duff, J.M. et al. Adequacy of Published Oncology Randomized Controlled Trials to Provide Therapeutic Details Needed for Clinical Application, JNCI: Journal of the National Cancer Institute, 2010; 102(10):702-705.
The Economist. Drug testing: Trials and Errors, The Economist, 25 July 2015. https://www.economist.com/news/ leaders/21659743-evidence-base-new-medicines-flawed-time-fix-it-trials-and-errors [Accessed: 08 November 2017]
European Medicines Agency. Posting of clinical trial summary results in European Clinical Trials Database (EudraCT) to become mandatory for sponsors as of 21 July 2014, European Medicines Agency, 2014. http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2014/06/news_detail_002127.jsp&mid=WC0b01ac058004d5c1 [Accessed 20 September 2017]
European Medicines Agency. New judicial decisions at odds with EMA’s efforts to allow access to documents on medicines, EMA appeals interim measures, European Medicines Agency. http://www.ema.europa.eu/docs/en_GB/ document_library/Press_release/2016/09/WC500213362.pdf [Accessed 26 September 2017]
European Ombudsman. Decision of the European Ombudsman closing his inquiry into complaint 2560/2007/ BEH against the European Medicines Agency, 2010.
European Union. Guidance on the information concerning paediatric clinical trials to be entered into the EU Database on Clinical Trials, Official Journal of the European Union, 2009; C 28/01. https://ec.europa.eu/health/ sites/health/files/files/eudralex/vol-10/2009_c28_01/2009_c28_01_en.pdf [Accessed 20 September 2017]
European Court of Justice. PTC Therapeutics International v EMA, no date [case in progress] http://curia.europa.eu/juris/document/document.jsf?text=&docid=188741&pageIndex=0&doclang=en&mode=lst&dir=&occ=first&part=1&cid=1247554 [Accessed 24 November 2017]
FDA Transparency Working Group. Blueprint for Transparency at the U.S. Food and Drug Administration, Recommendations to Advance the Development of Safe and Effective Medical Products, Journal of Law, Medicine & Ethics, 2017; 45(4).
Feuerstein, A. Biotech Stock Mailbag: Ionis, Trust Issues and ‘Oops! Don’t Look at That Slide Deck. The Street, 2016. https://www.thestreet.com/story/13665173/1/biotech-stock-mailbag-ionis-trust-issues-and-oops-don-t-look-at-that-slide-deck.html [Accessed 08 November 2017]
Glasziou, P. & Chalmers, I. Is 85% of health research really ‘wasted’? BMJ Opinion, 2016. http://blogs.bmj.com/ bmj/2016/01/14/paul-glasziou-and-iain-chalmers-is-85-of-health-research-really-wasted/ [Accessed: 27 September 2017]
Goldacre, B. Bad Pharma: How Drug Companies Mislead Doctors and Harm Patients, Fourth Estate, London, UK. 2012. ISBN: 978-0-00-735074-2.
Goldacre, B., Drysdale, H. & Heneghan, C. The Centre for Evidence Based Medicine Outcome Monitoring Project (COMPare) Protocol, CEBM, University of Oxford, 2016. http://compare-trials.org/wp-content/ uploads/2016/01/COMPare-Protocol-18.5.2016.pdf [Accessed 20 September 2017]
Goldacre, B., Drysdale, H. & Powell-Smith, A. et al. COMPare Trials Project. 2016. http://compare-trials.org/ [Accessed 20 September 2017]
Goldacre, B. et al. Pharmaceutical companies’ policies on access to trial data, results and methods: Audit study, BMJ, 2017; 358:j3334.
Golder, S. et al. Reporting of Adverse Events in Published and Unpublished Studies of Health Care Interventions: A Systematic Review, PLoS Medicine, 2016; 13(9).
Gøtzsche, P.C. & Jørgensen, A.W. Opening up data at the European Medicines Agency, BMJ, 2012;342.
Health Action International Europe. Protecting citizens’ health: Transparency of clinical trial data on medicines in the EU, HAI Europe, 2013.
Harriman S. & Patel J. When are clinical trials registered? An analysis of prospective versus retrospective registration, Trials, 2016. https://trialsjournal.biomedcentral.com/articles/10.1186/s13063-016-1310-8 [Accessed 20 September 2017]
Health Research Authority. Research registration and research project identifiers, 2017. https://www.hra.nhs.uk/planning-and-improving-research/research-planning/research-registration-and-research-project-identifiers/ [Accessed 23 November 2017].
Higgins, J.P.T. & Green, S. Cochrane Handbook for Systematic Reviews of Interventions, Version 5.1, The Cochrane Collaboration, 2011.
Hoffmann, T. et al. Focus on sharing individual patient data distracts from other ways of improving trial transparency, British Medical Association, 2017.
Hwang, T.J. et al. Failure of investigational drugs in late-stage clinical development and publication of trial results, JAMA Internal Medicine, 2016; 176(12):1826-1833.
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, http://www.ich.org/home.html [Accessed 20 September 2017]
Jefferson, T. et al. Oseltamivir for influenza in adults and children: systematic review of clinical study reports and summary of regulatory comments, BMJ, 2014; 348:g2545.
Jefferson, T. et al. Risk of Bias in industry-funded oseltamivir trials: comparison of core reports versus full clinical study reports, BMJ Open, 2014; 4:e005253.
Jones, P. et al. Comparison of Registered and Reported Outcomes in Randomized Clinical Trials Published in Anesthesiology Journals, Anesthesia and Analgesia, 2017; 125(4):1292- 1300.
Kiley, R. et al. Data Sharing from Clinical Trials – A Research Funder’s Perspective, New England Journal of Medicine, 2017; 377:1990-1992.
Koenig, F. et al. Sharing clinical trial data on patient level: opportunities and challenges, Biometrical Journal [Biometrische Zeitschrift], 2015, 57(1):8-26.
Kolstoe, S.E., Shanahan, D.R. & Wisely, J. Should research ethics committees police reporting bias? BMJ: British Medical Journal, 2017; 356.
Laine, C. et al. Update on Trials Registration: Clinical Trial Registration: Looking Back and Moving Ahead, International Committee of Medical Journal Editors, 2007. http://www.icmje.org/news-and-editorials/clincial_trial_reg_ jun2007.html [Accessed 20 September 2017]
The Lancet. Research: increasing value, reducing waste Series on research waste, The Lancet, 2014. www.thelancet.com/series/research [Accessed 28 September 2017]
Lexchin, J. et al. Pharmaceutical industry sponsorship and research outcome and quality: systematic review, BMJ, 2003; 326(7400):1167-1170.
Maruani, A. et al. Impact of sending email reminders of the legal requirement for posting results on ClinicalTrials. gov: cohort embedded pragmatic randomized controlled trial, BMJ, 2014; 349:g5579
Mayo-Wilson, E., Doshi, P. & Dickersin, K. Are manufacturers sharing data as promised? BMJ, 2015; 351:h4169.
Mayo-Wilson, E. et al. Practical guidance for using multiple data sources in systematic reviews and meta- analyses, Research Synthesis Methods, 2017; 10:1002/jrsm.1277.
McGauran, N. et al. Reporting bias in medical research – a narrative review. Trials, 2010; 11(1) 37.
Modjarrad, K. Developing Global Norms of Sharing Data and Results during Public Health Emergencies, PLoS Medicine, 2016; 13(1): e1001935.
Miller, J.E., Korn, D. & Ross, J.S. Clinical trial registration, reporting, publication and FDAAA compliance: a cross-sectional analysis and ranking of new drugs approved by the FDA in 2012, BMJ Open, 2015; 5(11).
Mooney, L. A. & Fay, L. Cross-sectional study of Pfizer-sponsored clinical trials: assessment of time to publication and publication history, BMJ Open 2016; 6:e012362.
Moscicki, R. Responsible Sharing of Clinical Trial Data: An FDA Perspective, US Food and Drug Administration, 2014. http://www.cbinet.com/sites/default/files/files/Moscicki_Richard_pres.pdf [Accessed 27 November 2017]
MRC. Good Practice Principles for Individual Participant Data from Publicly Funded Clinical Trials, 2015. www.methodologyhubs.mrc.ac.uk/files/7114/3682/3831/Datasharingguidance2015.pdf [Accessed 10 November 2017]
Nasser, M. et al. What are funders doing to minimise waste in research? The Lancet, 2017; 389(10073):1006- 1007.
Nguyen, T.A. et al. Public availability of results of trials assessing cancer drugs in the United States, Journal of Clinical Oncology, 2013; 31(24):2998-3003.
Onakpoya, I.J., Heneghan, C.J. & Aronson, J.K. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: a systematic review of the world literature, BMC medicine, 2016; 14(10).
Pica, N. & Bourgeois, F. Discontinuation and nonpublication of randomized clinical trials conducted in children. American Academy of Paediatrics, 2016: e20160223.
Piller, C. Failure to report: A STAT investigation of clinical trials reporting, STAT, 2015. https://www.statnews.com/2015/12/13/clinical-trials-investigation/ [Accessed 20 September 2017]
Piller, C. Interview with AllTrials, 2016. http://www.alltrials.net/news/fdaaa-enforcement-clinical-trials-fda-nih/ [Accessed 20 September 2017]
Pisani, E. & Botchway S. Sharing individual patient and parasite-level data through the WorldWide Antimalarial Resistance Network platform: A qualitative case study, Wellcome Open Research, 2017; 2:63.
Powell-Smith, A. et al. The TrialsTracker: automated ongoing monitoring of failure to share clinical trial results by all major companies and research institutions. F1000Res, 2016; 52629.
The PLoS Medicine Editors. Can Data Sharing Become the Path of Least Resistance? PLoS Medicine, 2016, 13(1):e1001949.
Prayle, A.P., Hurley, M.N. & Smyth, A.R. Compliance with mandatory reporting of clinical trial results on ClinicalTrials.gov: cross sectional study, BMJ, 2012; 344:d7373.
Public Accounts Committee. Thirty-Fifth Report, Access to clinical trial information and the stockpiling of Tamiflu, 2013. https://publications.parliament.uk/pa/cm201314/cmselect/cmpubacc/295/29502.htm [Accessed 26 September 2017]
Quintano, A. S. Hai Europe calls on EMA’s Management Board to stand for data transparency, Health Action International. http://haiweb.org/hai-europe-calls-on-emas-management-board-to-stand-for-data-transparency/ [Accessed 27 September 2017]
Ritchie, J. The European Medicines Agency Gives Unprecedented Public Access to Clinical Trial Data, Association of Medical Research Charities (AMRC), 2016. https://www.amrc.org.uk/blog/the-european-medicines- agency-gives-unprecedented-public-access-to-clinical-trial-data [Accessed 20 September 2017]
Riveros, C. et al. Timing and Completeness of Trial Results Posted at ClinicalTrials.gov and Published in Journals, PLoS Medicine, 2013; 10(12).
Rosati, P. et al. Major discrepancies between what clinical trial registries record and paediatric randomised controlled trials publish, Trials 2016; 17:430.
Ross, J. et al. Publication of NIH funded trials registered in ClinicalTrials.gov: cross sectional analysis, BMJ, 2012; 344:d7292.
Ross, J.S. Clinical research data sharing: What an open science world means for researchers involved in evidence synthesis, Systematic Reviews, 2016; 5:159.
Saini, P. et al., Selective reporting bias of harm outcomes within studies: findings from a cohort of systematic reviews, BMJ 2014; 349: g6501.
Schroll, J.B., Penninga E.L. & Gøtzsche, P.C. Assessment of Adverse Events in Protocols, Clinical Study Reports, and Published Papers of Trials of Orlistat: A Document Analysis, PLoS Medicine, 2016; 13(8):e1002101.
Smith, R. Should scientific fraud be a criminal offence? BMJ Opinion, 2013. http://blogs.bmj.com/bmj/2013/12/09/ richard-smith-should-scientific-fraud-be-a-criminal-offence/ [Accessed 26 September 2017]
Smithy, J.W., Downing, N.S. & Ross J.S. Publication of pivotal efficacy trials for novel therapeutic agents approved between 2005 and 2011: a cross-sectional study, JAMA Intern Med 2014;174(9):1518-20.
Song, F. et al. Dissemination and publication of research findings: an updated review of related biases, Health Technol Assess, 2010; 14(8): 1-193.
Song, F., Loke, Y. & Hooper, L. Why are medical and health-related studies not being published? A systematic review of reasons given by investigators, PLoS One, 2014; 9(10).
Southworth C. Study Registration Loopholes, Journal of Clinical Research Best Practice, 2011, 7(7).
Steinbrook, R. Gag Clauses in Clinical-Trial Agreements, New England Journal of Medicine, 2005; 352(21), 2160-2162.
Strom, B.L. et al. Data Sharing, Year 1 – Access to Data from Industry – Sponsored Clinical Trials, New England Journal of Medicine, 2014; 371:2052-2054.
Sydes, M.R. et al. Sharing data from clinical trials: the rationale for a controlled access approach, Trials, 2015; 16:104.
Taichman, D. et al. Sharing Clinical Trial Data, BMJ, 2016; 352:i255.
Taichman, D. Data Sharing Statements for Clinical Trials: A Requirement of the International Committee of Medical Journal Editors, Annals of Internal Medicine, 2016; 164:505-6.
Tang, E. et al. Comparison of serious adverse events posted at ClinicalTrials.gov and published in corresponding journal articles, BMC Medicine, 2015; 13:189.
Tierney, J.F. et al. How Individual participant data meta-analyses have influenced trial deign, conduct and analysis, Journal of Clinical Epidemiology, 2015; 68(11): 1325-1335.
Tierney, J.F. et al. Individual Participant Data (IPD) Meta-analyses of Randomized Controlled Trials: Guidance on Their Use, PLoS Medicine, 2015; 12(7).
Torjesen, I. European drug agency backtracks on plan to give researchers access to clinical trial reports, BMJ, 2014; 348:g3432.
Turdur-Smith, C. et al. How should Individual Participant Data (IDP) from publicly funded clinical trials be shared? BMC Medicine, 2015; 13:298.
Turner E.H. Posting FDA new drug application reviews, JAMA, 2007; 298:863-4.
Turner, E.H. et al. Selective Publication of Antidepressant Trials and Its Influence on Apparent Efficacy, New England Journal of Medicine, 2008, 358: 252-260.
Turner, E.H. How to access and process FDA drug approval packages for use in research, BMJ, 2013; 347:f5992
UNAIDS & World Health Organisation. Ethical considerations in biomedical HIV prevention trials, UNAIDS & WHO guidance document, 2012. http://www.unaids.org/sites/default/files/media_asset/jc1399_ethical_considerations_ en_0.pdf [Accessed 20 September 2017]
United Nations. Report of the United Nations Secretary-General’s High-Level Panel on Access to Medicines, Promoting innovation and access to health technologies, High-level Panel on Access to Medicines, 2016. http://www.politico.eu/wp-content/uploads/2016/09/HLP-Report-FINAL-Sept-2016.pdf [Accessed 20 September 2017]
U.S. Food & Drug Administration. What is a Serious Adverse Event? Reporting Serious Problems to the FDA, 2016. https://www.fda.gov/safety/medwatch/howtoreport/ucm053087.htm [Accessed 10 November 2017]
Wieseler, B. et al. Finding studies on Reboxetine: a tale of hide and seek, BMJ, 2010; 341:c4942.
Wieseler, B. et al. Access to regulatory data from the European Medicines Agency: the times they are a-changing, Systematic reviews, 2012; 1:50.
Wieseler, B. et al. Impact of document type on reporting quality of clinical drug trials: a comparison of registry reports, clinical study reports, and journal publications, BMJ, 2012; 344:d8141.
Wieseler, B. et al. Completeness of Reporting of Patient-Relevant Clinical Trial Outcomes: Comparison of Unpublished Clinical Study Reports with Publicly Available Data, PLoS Medicine, 2013 10(10).
Wieseler, B. Response to European drug agency backtracks on plan to give researchers access to clinical trial reports, BMJ, 2014; 348. http://www.bmj.com/content/348/bmj.g3432/rapid-responses [Accessed 10 November 2017]
Woloshin, S. et al. The Fate of FDA Postapproval Studies, New England Journal of Medicine, 2017; 377: 1114- 1117.
WHO. International Standards for Clinical Trial Registries, 2012. http://apps.who.int/iris/bitstream/10665/76705/1/9789241504294_eng.pdf [Accessed 20 September 2017].
WHO. Developing Global Norms for Sharing Data and Results during Public Health Emergencies, 2015. http://www.who.int/medicines/ebola-treatment/data-sharing_phe/en/ [Accessed 20 September 2017].
WHO. Statement on Public Disclosure of Clinical Trial Results, 2015. http://www.who.int/ictrp/results/reporting/en/ [Accessed 20 September 2017].
WHO. Joint statement on public disclosure of results from clinical trials, 2017. http://www.who.int/ictrp/results/ jointstatement/en/ [Accessed 05 October 2017]
World Medical Association. Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects, 2013. https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human- subjects/ [Accessed 20 September 2017]
Worldwide Antimalarial Resistance Network (WWARN) DP Study Group. The Effect of Dosing Regimens on the Antimalarial Efficacy of Dihydroartemisinin-Piperaquine: A Pooled Analysis of Individual Patient Data, PLoS Medicine, 2013; 10(12).
Zarin, D. et al. Issues in the Registration of Clinical Trials, JAMA, 2007; 297(19):2112-2120.
Zarin, D. & Tse, T. Moving Towards Transparency of Clinical Trials, Science, 2008; 319(5868): 1340–1342.
Zarin, D. et al. The ClinicalTrials.gov Results Database – Update and Key Issues, New England Journal of Medicine, 2011; 364:852-60.
Zarin, D. & Tse, T. Sharing Individual Participant Data (IDP) within the Context of the Trial Reporting System (TRS), PLoS Medicine, 2016; 13(1).
Zarin, D. et al. Update on Trial Registration 11 Years after the ICMJE Policy Was Established, New England Journal of Medicine, 2017; 376:383-391.