Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Dados financeiros do órgão encarregado de autorizar o uso de medicamentos na UE, dos quais a Investigate Europe teve acesso, mostram sua dependência em multinacionais como a Novartis, a Pfizer e a AstraZeneca.
A Agência Europeia de Medicamentos (EMA, siglas em inglês) é responsável por autorizar a comercialização de fármacos usados na Espanha e no resto da UE contra as principais doenças. Suas decisões têm um impacto direto sobre a conta resultados das empresas da setor. Ao mesmo tempo, um pequeno grupo de 21 multinacionais farmacêuticas contribui com quase metade dos mais de 400 milhões que o órgão regulador recebe anualmente.
A Investigate Europe revela, pela primeira vez, quem são as empresas que financiam a EMA. Os jornalistas obtiveram os dados por meio de uma solicitação de acesso a informações públicas de acordo com a legislação europeia sobre de transparência. Concretamente, eles pediram para saber a identidade e o valor pago por todas as empresas farmacêuticas em 2022. A EMA forneceu um arquivo com 41.640 pagamentos recebidos naquele ano, feitos por 3.564 entidades diferentes. A Agência cobra taxas (https://www.ema.europa.eu/en/about-us/fees-payable-european-medicines-agency) para solicitações de autorização de comercialização de fármacos e suas alterações, bem como taxas anuais para medicamentos autorizados.
A análise dos dados leva a uma conclusão principal: apesar de milhares de entidades pagarem taxas, um pequeno grupo de 21 multinacionais é responsável pela metade dos pagamentos. Em outras palavras: o orçamento da EMA depende em grande parte de um pequeno número de empresas, que são por sua vez, as principais beneficiárias das decisões que ela toma.
A Novartis está no topo da lista de pagamentos de 2022, com 19,6 milhões de euros, seguida pela Pfizer (14,3), depois pela AstraZeneca (12,5), pela Janssen (10,5), pela Roche (10,2) e pela GlaxoSmithKline (10,2). No total, as 21 multinacionais contribuíram com €165,4 milhões (veja a lista completa no gráfico do artigo original – link no cabeçalho).
Apesar de a EMA ter fornecido detalhes de todas o faturamento recebido, quando se entende que foi obrigada a fazê-lo pela legislação sobre transparência, ela censurou a identidade de alguns pagadores. Em uma carta aos jornalistas, a EMA explicou: “As informações comercialmente confidenciais relacionadas aos planos de desenvolvimento para o futuro foram editadas. Em particular, foram removidos os nomes de terceiros que pagaram honorários por consultoria científica para evitar que a divulgação do documento prejudicasse a proteção dos interesses comerciais de uma pessoa física ou jurídica, incluindo a propriedade intelectual”.
Além das taxas que recebe pelas autorizações de comercialização, a Agência também cobra das (https://www.ema.europa.eu/en/human-regulatory-overview/research-development/scientific-advice-protocol-assistance) empresas que solicitam, por assessoria científica enquanto elas desenvolvem um produto médico.
As informações censuradas são de pagamentos no valor de €36,2 milhões, o que representa 10% dos 364,2 milhões que a EMA faturou de empresas farmacêuticas em 2022. Portanto, a quantidade total paga pelas 21 multinacionais é ainda maior, pois elas têm o maior número de fármacos em desenvolvimento com assessoria científica da Agência.
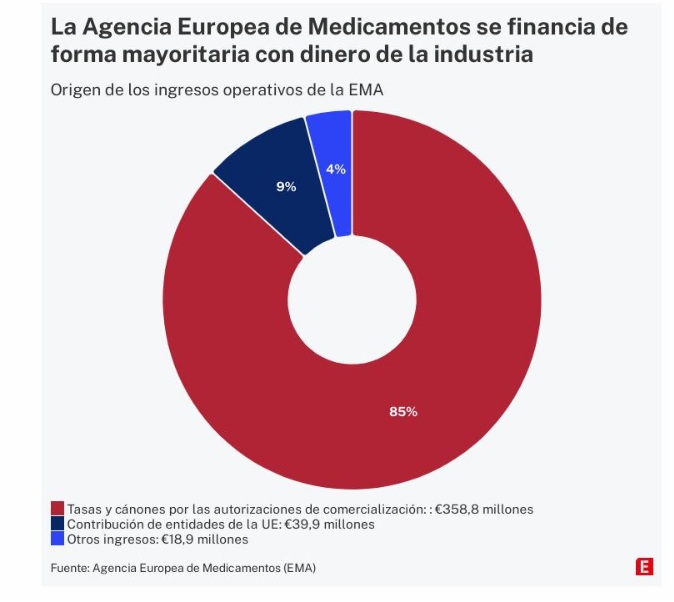
A dependência da EMA do dinheiro da indústria é quase total e tem aumentado constantemente nos últimos anos.
Quando foi criado em 1995, apenas 20% do financiamento vinha de empresas. O restante veio dos orçamentos da União Europeia. Em 2022, o dinheiro operacional da Agência totalizou 417 milhões, com pagamentos de farmacêuticas representando mais de 85% do total [nas contas anuais mostram receitas de 358,8 milhões, como está representado no gráfico, enquanto nas informações facilitada pela Transparência são 364,2, porque esse número inclui 5,4 milhões recebidos em 2022 de anos anteriores]. Em 2024, espera-se que mais de 90% sejam provenientes de taxas pagas pela indústria. A contribuição da UE vem sendo reduzida paralelamente e, em 2022, ficou abaixo de 10% das receitas.
O aumento das receitas procedentes das farmacêuticas se deve, em parte, ao fato de os processos de autorização terem disparado. O regulador europeu deu luz verde para uma média de 85 novos medicamentos por ano na última década, em comparação com uma média anual de 49 nos 15 anos anteriores.
É importante ressaltar que esse modelo, no qual as empresas farmacêuticas financiam o órgão que as regulamenta, também é aplicado nas agências estatais da maioria dos países europeus. Com uma grande exceção: a França. Noventa e dois por cento das receitasda Agência Nacional de Segurança de Medicamentos Francesa vêm da Previdência Social, por meio do seguro de doenças (assurance maladie). Tem por tanto, uma origem pública.
Já a Agência Espanhola de Medicamentos e Produtos de Saúde (AEMPS) teve um faturamento de € 83,2 milhões em 2022 , dos quais 68,9 foram taxas pagas pela indústria, o equivalente a 82,8% do total. Outros €11,7 milhões (14,0% do total) foram faturamentos de serviços prestados à EMA correspondentes a atividades de autorização e monitoramento de fármacos aprovados por procedimento centralizado.
A venda de um medicamento em um país da UE pode ser autorizada de três maneiras: pela EMA, no que é conhecido como procedimento centralizado; por Agências estatais, como a AEMPS; ou através do reconhecimento mútuo e do procedimento descentralizado, pelo qual um país aprova um fármaco já autorizado em outro Estado. Das 1.800 novas autorizações para medicamentos por ano na Espanha, 12% são autorizadas pela EMA, 33% pela AEMPS e 55% seguem procedimentos descentralizados ou de reconhecimento mútuo.
Porém, a EMA tem o monopólio da aprovação de todos os medicamentos para câncer, diabetes, doenças neurodegenerativas, doenças raras ou virais e os produzidos por meio de processos biotecnológicos ou por terapias genéticas. Assim sendo, embora em termos quantitativos menos fármacos sejam aprovados pela EMA, em termos qualitativos sua importância é muito maior. E naturalmente, também tem maior importância em termos econômicos para as empresas, que geram a grande maioria de seus lucros com fármacos aprovados pela EMA.
Para realizar os processos de avaliação, a EMA recorre a pesquisadores de agências estatais, o que explica por que 14% do faturamento da AEMPS espanhola procede da Agência Europeia.
Esse modelo de financiamento, tanto da EMA quanto da maioria das Agências estatais, é criticado por especialistas consultados pela Investigate Europe pelos potenciais conflitos de interesse. Os laços estreitos da EMA com a indústria são bem conhecidos, diz Yannis Natsis, que atuou em sua diretoria por dois anos e meio, representando os provedores de serviços de saúde. “A EMA tem uma longa tradição de estreita colaboração com as empresas que ela deve regulamentar”, diz Natsis.
O médico Fernando Lamata, um dos maiores especialistas da Espanha na questão de preços de medicamentos, acredita que “não deveria haver financiamento privado” nem da EMA nem das Agências estatais. A razão para isso é óbvia para ele: “No sistema atual, quem vai analisar os dados dos ensaios clínicos? E quem paga a EMA? A mesma indústria que realiza os estudos que a EMA terá que analisar”.
Na Agência Europeia de Medicamentos, eles veem isso de outra forma. Um representante da agência lembra que as autoridades européias já tomaram “uma decisão política em meados dos anos 90 para exigir que as empresas farmacêuticas contribuam com os custos da regulamentação e avaliação de medicamentos da EMA”. Em sua opinião, trata-se de uma questão de justiça para com os contribuintes: “Parece justo, levando em conta que as autorizações de comercialização trazem benefícios econômicos consideráveis para o solicitante (ou seja, acesso ao mercado único da UE), que o custo da avaliação científica e do monitoramento pós-autorização dos medicamentos seja compartilhado pelo solicitante e não suportado exclusivamente pelos contribuintes.” Caso contrário, acrescenta a representante, “as empresas se beneficiariam em dobro: primeiro, acessando o mercado da UE, onde podem ter beneficios, e, segundo, não pagando nenhum custo regulatório para acessá-lo”.
Além disso, a EMA nega que os pagamentos da indústria afetem as decisões que toma: “Os solicitantes pagam por um procedimento, mas não pelo resultado do mesmo. Isso significa que uma empresa paga no momento em que envia uma solicitação à EMA; em seguida, a Agência realiza uma avaliação independente. Usando a analogia de um exame de direção, é preciso pagar para fazer um exame de direção, mas não há garantia de aprovação no exame”.
A Farmaindustria, a organização de empresas espanholas, também não vê problema no fato de suas empresas pagarem à agência que as regulamenta: “A lei estabelece taxas para as empresas farmacêuticas pelos serviços prestados pela AEMPS, como acontece com muitos serviços públicos em que o usuário paga taxas justamente pelo uso desses serviços. Esse pagamento de taxas não gera nenhum tipo de conflito de interesses, como é óbvio”. De qualquer forma, acrescenta o representante, “a indústria não seria indiferente à abolição das taxas da AEMPS, que ainda são um custo adicional para as empresas”.
Alguns membros da comunidade científica estão preocupados com o fato de a EMA estar autorizando o uso de fármacos com ensaios clínicos pouco exigentes sobre eficácia e segurança. “A EMA está aprovando novos medicamentos mais rapidamente e com menos dados clínicos disponíveis. Cada vez é mais difícil avaliar o real benefício adicional em comparação com os medicamentos existentes”, diz Beate Wiseler, do prestigiado instituto IQWiG, responsável pela qualidade e eficiência sanitária na Alemanha.
Essa preocupação é desencadeada entre os especialistas quando analisam os procedimentos especiais que a EMA tem para garantir o acesso rápido ao mercado para determinados fármacos. Existem três rotas rápidas para a obtenção de permissão: autorização condicional de comercialização (CMA, siglas em inglês), avaliação acelerada e circunstâncias excepcionais.
A Investigate Europe analisou todos os fármacos aprovados pela EMA desde 2004, quando o primeiro desses procedimentos especiais foi introduzido, até dezembro de 2023. No total, 198 medicamentos entraram no mercado por meio de uma das três vias aceleradas. Desses, 173 ainda estão sendo comercializados, 16 foram retirados pelos laboratórios, em 7 casos a licença expirou e apenas duas vezes a Agência revogou a autorização (Lartruvo (https://www.aemps.gob.es/informa/notasinformativas/medicamentosusohumano-3/2019-muh/actualizacion-de-la-informacion-sobre-olaratumab-%E2%96%BClartruvo-recomendacion-de-retirada-de-la-autorizacion-de-comercializacion/), patenteado pela Eli Lilly e usado para o tratamento de determinados sarcomas, e Adakveo (https://www.aemps.gob.es/informa/la-aemps-actualiza-la-informacion-sobre-crizanlizumab-adakveo-recomendacion-de-retirada-de-la-autorizacion-de-comercializacion/), de propriedade da Novartis e autorizado a prevenir crises vaso-oclusivas).
Um representante da EMA destacou que, desde 2004, a Agência autorizou “mais de 1.400 medicamentos”, de modo que os aprovados por meio de uma das vias aceleradas representam 14%.
A via mais utilizada, e a que gera a maior atenção crítica da comunidade científica, é a autorização condicional de comercialização. Essa é concedida com informações insuficientes sobre a eficácia ou a segurança do medicamento, para atender a “necessidades médicas não atendidas”, com a condição de que a empresa forneça posteriormente as evidências científicas que faltam. Se isso for feito, o medicamento obterá uma autorização de comercialização padrão.
A análise da Investigate Europe constatou três fatos impressionantes: que o uso desse procedimento se multiplicou nos últimos cinco anos; que dois terços dos medicamentos aprovados por essa via pertencem ao grupo de 21 multinacionais que sugam o financiamento da EMA, e que pode levar até dez anos para que os laboratórios apresentem as evidências científicas que faltam.
A Agência concedeu a primeira autorização condicional em 2006 e, até o final de 2023, aprovou um total de 91 medicamentos. 51 deles receberam aprovação nos últimos cinco anos, enquanto os 13 anos anteriores somente foram aprovados 40. Quando questionada sobre esses números, a representante da EMA ressaltou que o uso desse procedimento para aprovar as vacinas contra a Covid-19 poderia explicar parcialmente esses números. Porém, somente sete medicamentos relacionados à Covid-19 receberam esse tipo de autorização. Portanto, mesmo sem levar esses sete fármacos em conta, a conclusão é a mesma: entre 2019 e 2023, a Agência usou essa via especial em mais ocasiões do que nos 13 anos anteriores.
Os dados mostram também com clareza que os principais beneficiários da autorização condicional são precisamente os grandes pagadores da EMA. Do grupo de 21 multinacionais, três não contam com uma autorização deste tipo (Novo Nordisk, Accord e Teva). As outras 18 têm um total de 61 autorizações condicionais, ou seja, 67% do total. A Janssen é a que tem mais autorizações (9), seguida pela Novartis (7), pela Roche (7), pela Pfizer (6) e pela AstraZeneca (5).
Naturalmente, estas 21 multinacionais são também as que têm mais medicamentos no mercado, pelo que seria lógico pensar que seriam responsáveis por duas em cada três aprovações condicionais. Não é assim. Desde o início do funcionamento, em 1995, a EMA autorizou mais de 1700 fármacos, dos quais pouco mais de 800 pertencem às 21 multinacionais. Ou seja, 47%.
Em terceiro lugar, destaca o tempo que ocasionalmente os laboratórios demoram para fornecer provas de que o seu produto é eficaz e seguro. A autorização é concedida por um ano e pode ser renovada pelo mesmo período. Em 2022, a EMA concedeu a licença normal a dois medicamentos que tinham sido autorizados condicionalmente desde 2012 (Caprelsa e Adcetris) e a outro que estava no mercado desde 2013 (Bosulif). Hoje, cinco medicamentos que receberam autorização condicional em 2014 (Sirturo, Deltyba e Translarna) e 2016 (Ocaliva e uma vacina contra a gripe) ainda estão no mercado.
Questionada pelos jornalistas, a representante da EMA afirmou que o tempo médio que um medicamento com autorização condicional demora a obter a padronização “é de três anos e oito meses”. Mas os tempos médios nem sempre fornecem as informações mais relevantes. Investigadores do King’s College London descobriram que, entre 2013 e 2018, em metade dos casos, as provas exigidas não tinham sido fornecidas mais de sete anos após a autorização condicional. “Durante 30 anos, foi dito a nós que os estudos pós-comercialização preencheriam as brechas. Mas não foi o caso. Não recebemos essas provas”, lamenta Courtney Davis, socióloga médica da universidade britânica.
A Agência também defende a sua política em matéria de autorizações condicionais, que “só são recomendadas quando a relação risco-benefício do medicamento é positiva, o solicitante pode fornecer dados completos após a autorização, o fármaco responde a uma necessidade médica não satisfeita e o benefício da disponibilidade imediata para os pacientes supera os riscos inerentes ao facto de ainda serem requeridos dados adicionais”.
Precisamente o Ocaliva e o Translarna, dois dos fármacos que estão no mercado há mais tempo com uma autorização condicional, estão agora sendo questionados.
O Ocaliva é um tratamento para a cirrose biliar primária, uma doença autoimune do fígado. Em outubro de 2023, a EMA iniciou “uma revisão risco-benefício” do fármaco, “impulsionada pelos resultados finais dos dois estudos em doentes” que tinha solicitado em 2016, como parte dos requisitos para a concessão da autorização condicional. Em outras palavras, os resultados finais precisaram de sete anos para chegar. E não eram exatamente positivos. Um dos estudos, explica a representante da EMA, “não mostrou que o Ocaliva fosse mais eficaz do que o placebo em termos do número de doentes cuja doença se agravou ou que morreram. Além disso, os efeitos secundários, incluindo os efeitos secundários graves, foram mais frequentes nos doentes tratados com Ocaliva”. A Agência está analisando estes resultados, “juntamente com todos os outros dados disponíveis”, antes de decidir se vai retirar a autorização da Advanz Pharma.
A Comissão Europeia, com base nas recomendações da EMA, é quem toma formalmente decisões sobre os fármacos. Questionado pela Investigate Europe, um funcionário autorizado explicou que a Comissão “está ciente das preocupações relacionadas com o Ocaliva” e que o relatório da EMA é esperado em junho. “Assim que a Comissão receber este relatório, tomaremos as medidas regulamentares adequadas no que diz respeito à autorização deste produto”, concluiu.
O Translarna, por outro lado, é um medicamento para a distrofia muscular de Duchenne patenteado pela PTC Therapeutics. No momento da renovação anual em 2023, a EMA decidiu que este medicamento deveria ser retirado do mercado. Os efeitos secundários relatados incluem problemas cardíacos graves. A Comissão Europeia, que quase sempre segue as recomendações da EMA, não o fez nesta ocasião. Na resposta que o funcionário ofereceu à Investigate Europe, há ainda mais indícios de um conflito de interesses neste procedimento: “A Comissão pensa que a Agência deve considerar as possíveis implicações da recente sentença do Tribunal de Justiça da União Europeia (caso Hopveus), que afeta a composição dos painéis científicos consultivos que participam em uma avaliação e a sua conformidade com o princípio da imparcialidade objetiva.” O funcionário recusous fornecer mais informações quando questionado pelos jornalistas.
O caso Hopveus diz respeito a um fármaco com o mesmo nome, indicado para tratar a síndrome de abstinência alcoólica. A EMA recusou a autorização e o fabricante, a empresa francesa D&A Pharma, recorreu aos tribunais europeus. O TJUE, em uma sentença proferida em março, deu razão a eles e anulou a decisão negativa da EMA, detectando um conflito de interesses em um dos especialistas consultados.
Os conflitos de interesses dos especialistas são, sem dúvida, outro arquivo na Agência Europeia de Medicamentos. Para o Hopveus, para o Translarna e para outros casos.