Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
El 18 de diciembre de 2019, la Agencia Europa del Medicamento (AEM) dio el visto bueno a la comercialización de un preparado de esketamina (enantiómero S de la ketamina racémica), denominado Spravato® de la compañía farmacéutica Janssen-Cilag. El medicamento ha sido objeto de un considerable despliegue mediático destinado al público en general, en forma de artículos periodísticos sobre avances en salud mental y basados en opiniones de “expertos”, motivo por el que hemos decidido analizar el valor real del mismo. Ninguno de los autores tiene conflictos de interés asociados con los fabricantes ni con los que comercializan el producto o con sus competidores actuales o potenciales, ni con agencias y compañías de seguros médicos responsables de la financiación de los mismos. La información de este artículo se basa fundamentalmente en la ficha técnica aprobada por la AEM [1]. La sustancia se presenta en un dispositivo para administración nasal, que provee dos pulverizaciones cada una de 14mg, un total de 28mg de esketamina base por dispositivo, en solución acuosa. El preparado no requiere condiciones especiales de conservación y su periodo de validez es de 3 años. Esketamina nasal se considera que es un psicoanaléptico, incluido en la categoría de “otros antidepresivos” (grupo N06AX), la DDD se ha cuantificado en 8mg (56mg semanales) [2].
Los enantiómeros son moléculas idénticas salvo que una es imagen especular de la otra, se diferencian en la capacidad de rotar la luz polarizada y en la actividad biológica, siendo solo activo uno de ellos. La mezcla racémica contiene ambos enantiómeros (Ver Figura 1). La separación del enantiómero activo de la mezcla racémica es una de las estrategias empleadas por el sistema de patentes para prolongar la exclusividad en el mercado [3].
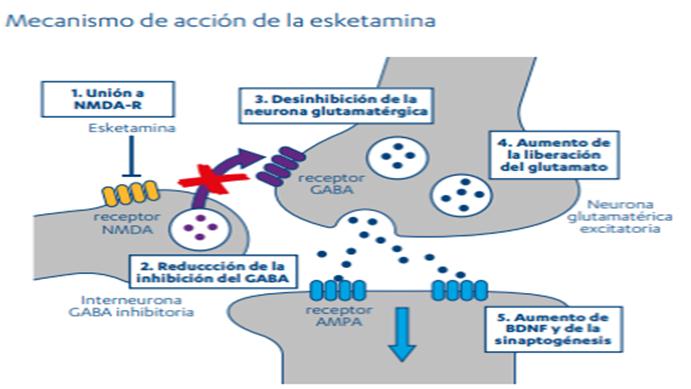
Esketamina es un antagonista no selectivo y no competitivo de los receptores NMDA (N-metil-D-aspartato), receptor ionotrópico de glutamato. El bloqueo del receptor NMDA conduce a un aumento transitorio de la liberación de glutamato que estimula los receptores AMPA (α-amino-3-hidroxi-5-metil-4-isoxazolpropionico ácido). Se supone que esto último causa un aumento de la señalización neurotrófica que puede contribuir al restablecimiento de la función sináptica en las regiones implicadas en la regulación del ánimo y el comportamiento emocional. El restablecimiento de la función dopaminérgica en las zonas implicadas en la motivación y la recompensa y la disminución de la estimulación en las zonas implicadas de la anhedonia pueden contribuir a la rápida respuesta. Ver Figura 2.
Las indicaciones aprobadas son:
En el tratamiento de la depresión mayor resistente a antidepresivos. Se ha estudiado en 5 ensayos clínicos fase III en pacientes adultos (18-86 años) con depresión mayor (DSM-5) resistente al tratamiento (que no respondían a dos antidepresivos orales a dosis y duración adecuadas en el episodio actual). Tres de los ensayos fueron a corto plazo y uno a largo plazo. Se incluyeron 1833 adultos, de los que 1601 se expusieron a esketamina nasal.
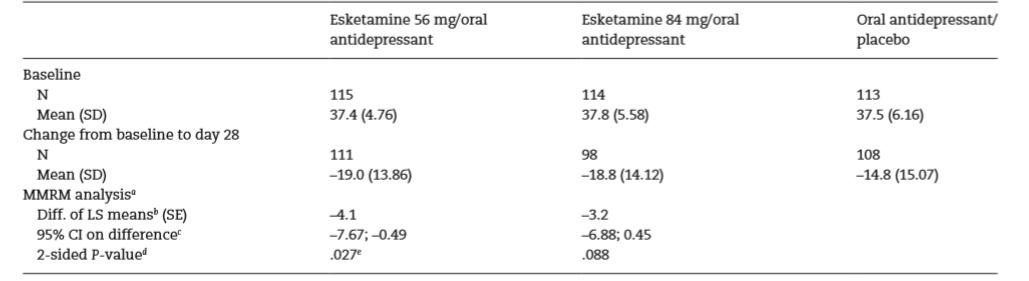
Los estudios a corto plazo se conocen como Transform 1, 2 y 3, (respectivamente TRD3001, TRD3002 y TRD3005) [4-6]. Tuvieron como objetivo valorar la eficacia de la adición de esketamina nasal, añadida a un antidepresivo oral de nueva instauración (venlafaxina, duloxetina, escitalopram o sertralina) como tratamiento de la depresión mayor resistente a dos fármacos antidepresivos orales en el episodio actual. Los tres tuvieron una duración de 4 semanas, fueron aleatorizados, de doble ciego y controlados con antidepresivo de nueva instauración, con uso de placebo nasal con aditivo amargo en la rama control para mantener el cegado. Los ensayos Transform 1 y 2 se realizaron en adultos entre 18 y <65 años, mientras que Transform 3 se realizó en ≥65 años. La dosis inicial de esketamina para <65 años fue 56mg, mientras que en ≥65 años fue de 28mg; posteriormente y según tolerancia, fueron de 56 a 84mg administradas 2 veces por semana durante 4 semanas (periodo denominado fase de inducción). En el estudio Transform 1 los sujetos fueron aleatorizados a una de 3 ramas de tratamiento, esketamina 56mg, esketamina 84mg o placebo, y las dosis se mantuvieron fijas. En los otros dos, Transform 2 y Transform 3, la dosis de cada sujeto fue flexible, en función de la respuesta, manteniéndolas entre 56 y 84mg de esketamina. El tratamiento con antidepresivo oral se mantuvo abierto desde el primer día. La variable principal de eficacia fue el cambio en la escala Montgomery-Asberg Depression Scale (MADRS) desde el valor basal al día 28 (esto es al final de la fase de inducción). La puntuación de la MADRS varía entre 0 y 60, un valor mayor indica una condición depresiva más grave. Un cambio negativo en esta puntuación indica mejoría de los síntomas, y una diferencia negativa entre el grupo con esketamina y grupo placebo favorece a la esketamina. A continuación, se muestran las características de los participantes en los ensayos Transform 1, 2 y 3. Ver Cuadro 1
El Cuadro 2 muestra la variable resultado principal en los estudios Transform 1, 2 y 3, el cambio en las puntuaciones MADRS desde el valor basal al final de la 4º semana de tratamiento.
La rama de esketamina a dosis fija de 56mg del estudio Transform 1, y la rama con esketamina a dosis flexibles entre 56 y 84mg del estudio Transform 2, mostraron resultados estadísticamente significativos superiores a las respectivas ramas placebo. En ambos casos la significación clínica es muy dudosa ya que no llegan a 5 unidades de 60 en la escala MADRS. Un descenso de 8 puntos en la MADRS puntuada por un clínico (o de 5 puntos si es autopuntuada) equivale a una mejoría mínima en la impresión clínica global [7]. Los intervalos de confianza al 95% de las diferencias de la rama de esketamina a dosis de 84mg en el ensayo Transform 1 y de la rama con esketamina a dosis flexible en el ensayo Transform 3 (≥65 años) con las respectivas ramas placebo incluyen el valor nulo, es decir no pudieron detectar que hubiera diferencia entre esketamina y placebo. Es más, el valor máximo de tales intervalos es <7, no alcanza una diferencia mínima clínicamente significativa.
Las tasas de respuesta y de remisión del trastorno depresivo mayor se establecieron como variables secundarias de resultado en los estudios Transform 1, 2 y 3. Se definió respuesta como una reducción ≥50% en la puntuación MADRS respecto del valor basal, al final de la fase de inducción. Remisión se definió como una reducción de la puntuación de la MADRS hasta un valor ≤12. El Cuadro 3 indica los valores que tomaron estas variables de resultados en los momentos preespecificados.
En el ensayo Trasnform 1 las tasas de respuesta en la rama con esketamina 56mg fueron superiores a las de la rama placebo en los 5 momentos evaluados. En este estudio las tasas la rama con esketamina 82mg solo se mostraron superiores a placebo en la 1º y 4º semana. Ninguna de las dos ramas con esketamina proporcionó tasas de remisión significativamente superiores a la del grupo placebo. En el ensayo Transform 2 la rama con esketamina nasal a dosis flexibles obtuvo tasas de remisión estadísticamente superiores a las de la rama placebo, aunque no hay diferencias entre rama activa y rama placebo en cuanto las tasas de respuesta en ninguno de los 5 momentos evaluados, salvo en la 3º semana. El ensayo Transform 3 no proporcionó diferencias entre la rama con esketamina y la rama con placebo en las tasas de respuesta en ninguno de los 5 momentos evaluados, ni en la tasa de remisión. Los datos dicotomizados (respondedores/no respondedores, remisión/no remisión) a partir de datos continuos (puntuación de los síntomas con la MADRS), como ocurre en estos estudios, constituyen una redundancia que trata de magnificar un efecto que previamente se había mostrado clínicamente irrelevante. Realmente son clínicamente indistinguibles los sujetos que puntúan justo debajo del punto de corte de los situados justo por encima del mismo [8].
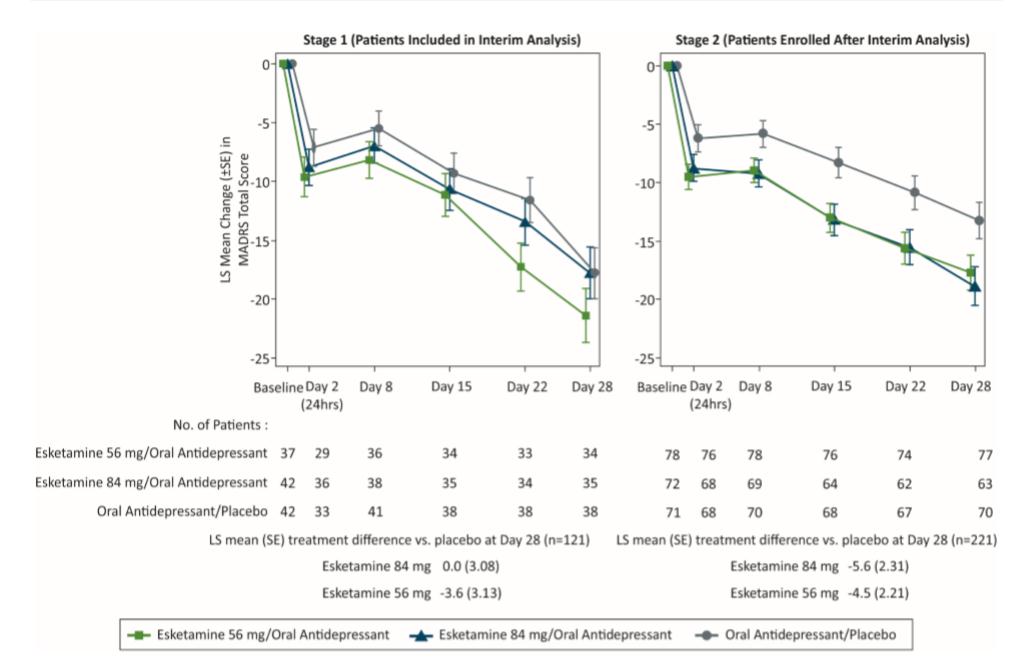
Hay 2 aspectos adicionales del estudio Tansform 1 que se deben comentar: el análisis estadístico secuencial y el análisis intermedio preespecificado en su protocolo [4]. El primero especificó que, para cada resultado objetivo, el examen de la significación del grupo con dosis de 56mg de esketamina nasal se llevaría a cabo solo si el grupo con dosis de 84mg se había mostrado significativamente superior al grupo placebo. Como no se cumplió la condición impuesta al grupo de 84mg, el grupo con 56mg no pudo ser analizado formalmente frente al placebo nasal, y los valores indicados deben ser considerados un análisis postrero, con un valor de generador de hipótesis, que no de rechazo de la hipótesis nula de ausencia de diferencias, ni se puede usar a efectos de obtención de la autorización de comercialización. Referente al análisis intermedio hay que decir que el comportamiento de los grupos a lo largo del tratamiento varió notablemente según los sujetos hubieran sido reclutados antes o después del mismo, acrecentándose las diferencias. Ver Figura 3A, que muestra el cambio medio (y desviación normal) de la puntuación total de la MADRS a lo largo del tiempo. En el escenario 1 se muestran los sujetos incluidos en el análisis intermedio, mientras que en el escenario 2 se incluyen los sujetos reclutados después del mismo. Este diferente comportamiento perfectamente puede estar indicando un escape del ciego, aunque los redactores del informe sobre el mismo lo niegan, favoreciendo las puntuaciones MADRS en los grupos con esketamina y desfavoreciendo al grupo con placebo. En el estudio Transform 3 también se realizó un análisis intermedio, tras el cual volvió a producirse un cambio de tendencia, ver figura 3B [6].
En el estudio Transform 2 también se preespecificó un análisis secuencial [5], en el que jerárquicamente se condiciona el análisis del porcentaje de remisión a que hubiera resultado significativa la diferencia de porcentajes de respuestas, situación que no se produce. Por tanto, la significación de la diferencia entre grupo con esketamina y grupo placebo de los porcentajes de sujetos en remisión, no tiene valor de rechazo de la hipótesis nula.
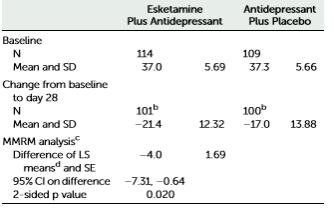
Otro aspecto a señalar es la discrepancia entre los valores de las variables de resultado de los estudios Transform 1 y Transform 2 indicados en la ficha técnica y en los informes publicados en las revistas profesionales [4,5]. En las publicaciones profesionales se muestran valores de eficacia superiores a los indicados por la AEM. Pueden compararse los valores mostrados en los Cuadros 2 y 3, procedentes de la ficha técnica, con los indicados en las Figuras 4A y 4B, tomadas de las respectivas publicaciones.
También se realizó un estudio a corto plazo en población japonesa (TRD2005) [9] con diseño de ramas paralelas, a doble ciego, de 4 semanas de duración (fase de inducción), con cuatro ramas de tratamiento a dosis fijas; placebo nasal, esketamina nasal 28mg, esketamina nasal 56mg y esketamina nasal 85mg, con un esquema 2:1:1:1, que incluyó 202 sujetos. Todos los sujetos habían mostrado ser resistentes a ≥1 pero <5 antidepresivos orales y recibieron durante todo el periodo un antidepresivo oral de nueva instauración. La variable resultado principal fue el cambio en la puntuación MADRS desde el momento basal al día 28. Ninguna de las ramas de tratamiento activo se mostró estadísticamente diferente al placebo. Ver Cuadro 4.
Se realizó un estudio a largo plazo, denominado Sustain 1 (TRD3003) [10], multicéntrico, aleatorizado, con control activo, a doble ciego con grupos paralelos, en el que la variable resultado principal fue “el tiempo hasta recaída tras aleatorización”. Parte de los sujetos procedían de los estudios Transform 1 y 2 (pacientes transferidos) y parte re reclutaron directamente para este estudio (pacientes de entrada directa). Tuvo 5 fases, 1) una inicial abierta para cribado de 4 semanas, 2) fase de inducción abierta de 4 semanas (estas dos fases fueron exclusivas para pacientes de entrada directa), 3) fase de optimización de 12 semanas (abierta para sujetos de entrada directa o a doble ciego para los transferidos), 4) fase de mantenimiento a doble ciego con retirada aleatorizada de esketamina y duración variable hasta evento resultado o finalización del estudio y 5) fase de seguimiento postratamiento.
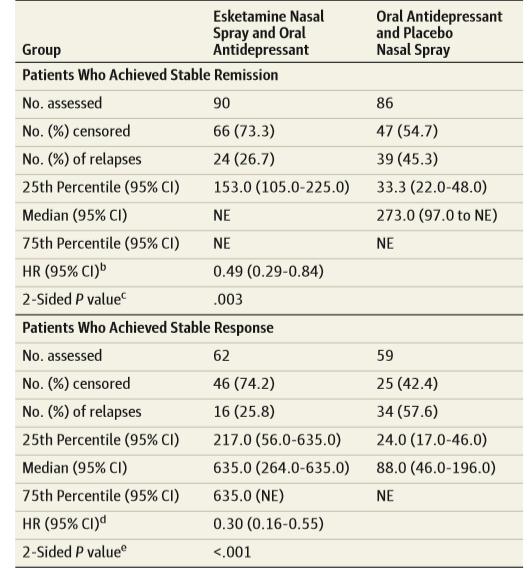
En la fase de optimización se incluyeron 705 sujetos, 150 y 118 procedieron respectivamente de las ramas activas de los estudios Transform 1 y Transform 2, y 437 fueron reclutados directamente. Estos últimos recibieron esketamina nasal de forma abierta por un periodo de inducción de 4 semanas (1º día 56mg, luego 56 u 84mg dos veces por semana) más antidepresivo oral. Al final de la fase de inducción abierta el 52% de los sujetos habían alcanzado la remisión (MADRS ≤12) y el 66% mostraron respuesta (reducción de la puntación MADRS ≥50% respecto al valor basal). Los 455 sujetos con respuesta entraron en la fase de optimización con dosis cada dos semanas si la puntuación MADRS se mantenía ≤12; o semanalmente durante las 4 semanas siguientes si esta tomaba un valor >12. Al final de esta fase de optimización, final de la semana 16 de tratamiento, 297 sujetos considerados estables fueron aleatorizados a continuar de forma doble ciego con esketamina nasal o a cambiarlo por placebo nasal, fase de mantenimiento. En remisión estable 176 sujetos (con puntuación total de la MADRS ≤ 12 en al menos 3 de las 4 últimas semanas de la fase de optimización) y otros 121 tenían respuesta estable (reducción ≥ 50 % de la puntuación total de la MADRS respecto al valor basal durante las 2 últimas semanas de la fase de optimización, pero sin alcanzar la remisión).
Se definió la recaída como una puntuación total de la MADRS ≥ 22 durante 2 semanas consecutivas o la hospitalización por empeoramiento de la depresión o cualquier otro acontecimiento de importancia clínica indicativo de recaída. La fase de mantenimiento fue de duración variable y continuó hasta que el paciente sufriera una recaída de los síntomas depresivos o se retirara por cualquier otro motivo, o se concluyó el estudio porque se había producido el número necesario de episodios de recaída. Todos los pacientes recibieron durante todo el tiempo un antidepresivo oral. La distribución del tiempo hasta la recaída durante la fase de mantenimiento entre los sujetos que alcanzaron la remisión se consideró la variable primaria final de eficacia, mientras el tiempo hasta la recaída entre los sujetos que tuvieron respuesta (sin remisión) se consideró variable final secundaria.
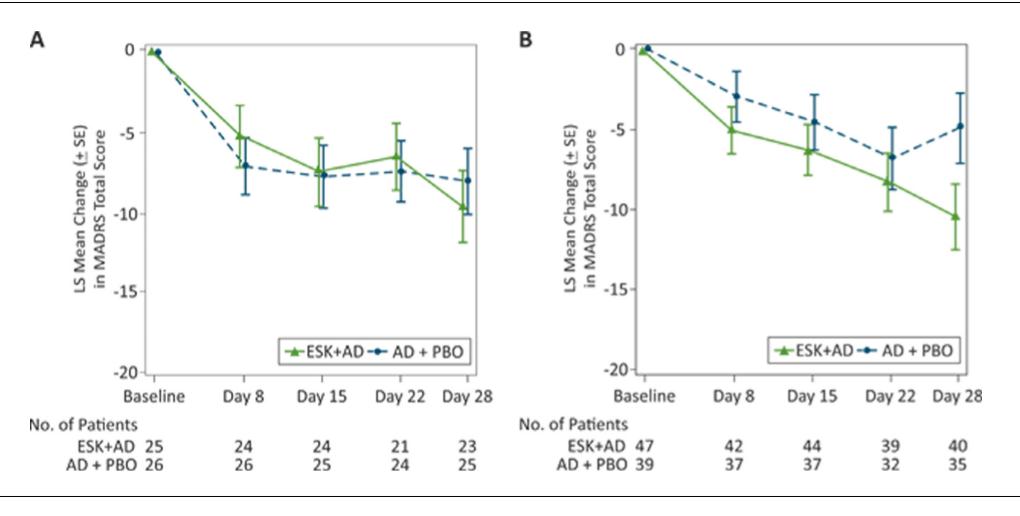
La figura 5 [10] muestra el tiempo hasta la recaída y el número de sujetos que se mantuvieron libres de recaída durante la fase de mantenimiento. Para los sujetos con remisión estable, la mediana del tiempo hasta la recaída en el grupo con placebo nasal (o más adecuadamente, a los que se les retiró esketamina) fue 273 días, mientras que en el grupo de continuación con esketamina no se pudo calcular la mediana de recaídas, ya que nunca alcanzó una tasa de recaída del 50% durante la fase de mantenimiento. Entre los pacientes con respuesta (sin remisión) que recibieron placebo (o más adecuadamente, a los que se les retiró la esketamina) la mediana de tiempo hasta la recaída fue de 88 días, frente a 635 días en el grupo que continuó con esketamina. Las diferencias fueron estadísticamente significativas.
En la semana 12 de mantenimiento a doble ciego la tasa de recaídas en el grupo esketamina fue de 13%, mientras que en el grupo placebo fue del 37% (la diferencia fue estadísticamente significativa, p<0,001, calculada por los comentaristas); a la semana 24 estos porcentajes fueron 32% y 46% respectivamente (diferencia que no alcanzó la significación estadística, p>0,05, calculada por los comentaristas). En la semana 12 de mantenimiento a doble ciego la tasa de recaídas en el grupo esketamina fue de 21%, mientras que en el grupo placebo fue del 47%; y a la semana 24 estos porcentajes fueron 21% y 56% respectivamente (diferencias estadísticamente significativas, respectivamente p<0,0027 y p<0,001, calculada por los comentaristas). Puede observarse en los Figura 6 A y B que la gran diferencia entre ambos grupos se produce inmediatamente tras la aleatorización, lo que hace pensar más bien en un problema de retirada.
Figura 6. Tiempo hasta recaída de los pacientes estables en el estudio Sustain 1(TRD3003) [10]
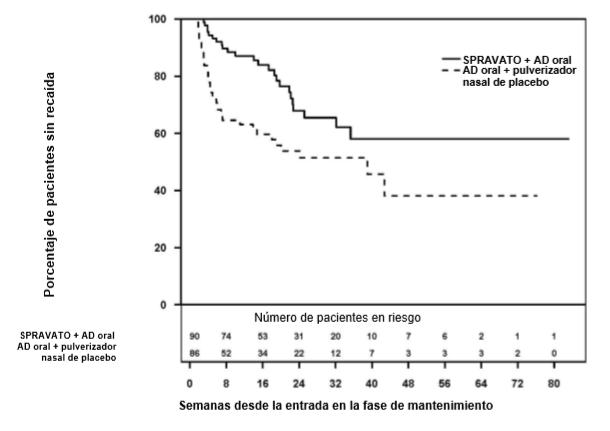
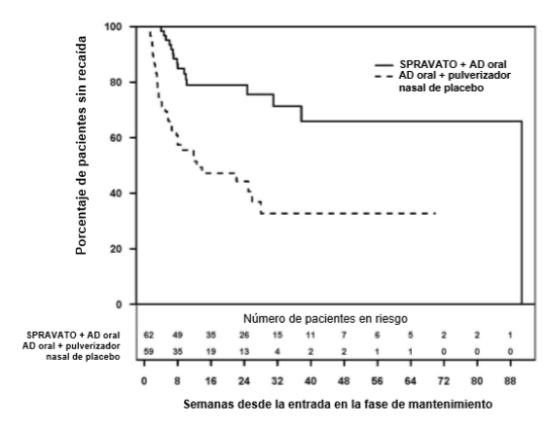
Los autores del estudio Sustain 1 concluyen que el mantenimiento con esketamina tras la respuesta antidepresiva previene eficazmente las recaídas. Nuestra conclusión es que tras la administración continua de esketamina, su retirada induce las recaídas depresivas. Considerar el fin del estudio cuando se han alcanzado un determinado número de recaídas favorece a la rama de continuación con un fármaco que cause dependencia, ya que la mayoría de estas se producirán en la rama en la que se retira el fármaco [11].
Tratamiento de la urgencia psiquiátrica debida a depresión mayor. También se ha estudiado en 2 ensayos clínicos: Aspire I (SUI3001) [12] y Aspire II (SUI 3002) [13], de fase III, en pacientes adultos (18-65 años), con trastorno depresivo mayor de moderado a grave (puntuación MADRS >28) y respuesta afirmativa a las preguntas B3 o B10 de la minientrevista neuropsiquiátrica internacional (“¿pensamiento [incluso momentáneamente] de dañarse o de herirse o de lesionarse a uno mismo: con al menos alguna intencionalidad o consciencia de que como resultado puede llegar a morir; o pensamiento de suicidio [es decir, de suicidarse]?”, “¿intencionalidad de actuar bajo pensamientos de suicidarse en las últimas 24 horas?” respectivamente). Incluyeron 456 pacientes adultos, de los que 227 se expusieron a esketamina nasal. Los dos ensayos tuvieron diseño similar, fueron con asignación aleatorizada, realizados a doble ciego, controlados con placebo y multicéntricos. En ambos estudios los sujetos recibieron esketamina nasal 84mg (que se podía bajar una única vez a 56mg si no se toleraba la dosis anterior) o placebo nasal dos veces por semana; todos los sujetos recibieron tratamiento de referencia integral (incluyendo diversas modalidades de uso de antidepresivos) para la emergencia por trastorno depresivo y hospitalización, esta última justificada por el riesgo inmediato de suicidio.
Las características de los sujetos asignados al grupo con esketamina y los asignados al grupo placebo no se diferenciaron en ninguno de los dos estudios. La edad mediana de los participantes fue 40 años (intervalo 18-64), el 61 % eran mujeres; el 73 % caucásicos y el 6 % de raza negra. El 63 % de los pacientes había intentado suicidarse al menos una vez, el 92% había recibido terapia antidepresiva previa a la inclusión en el estudio. Como tratamiento de referencia durante el ensayo, el 40% recibió antidepresivo en monoterapia, 52% antidepresivo más potenciación y el 6% ambas modalidades. La variable principal de resultado de eficacia fue el cambio en la puntuación MADRS a las 24h de la primera dosis (el día 2) respecto a la puntuación basal. La eficacia relacionada con la ideación y la conducta suicida fue evaluada empleando la herramienta de evaluación de ideación y conducta suicidas (SIBAT), siendo la segunda variable principal el cambio en la escala revisada de impresión clínica global de la gravedad de la suicidabilidad (CGI-SS-r). Ambos estudios se analizaron conjuntamente en otra publicación [14], los datos indicados en las publicaciones difieren ligeramente de los indicados en la ficha técnica aprobada por la AEM, que son los que se muestran en la Cuadro 5.
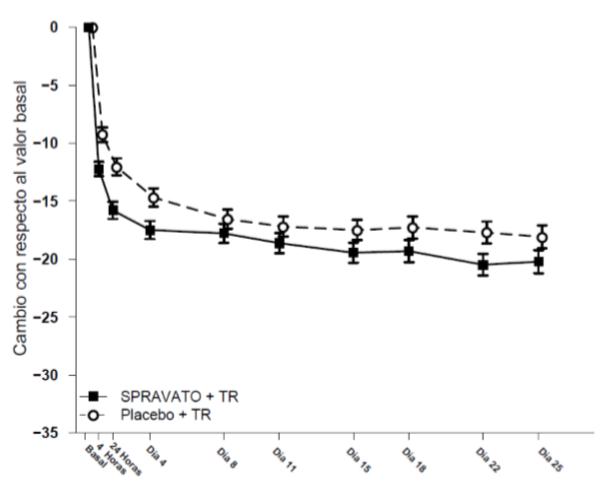
En los dos estudios SUI [12, 13] esketamina nasal redujo la puntuación SIBAT más que el placebo, ambos además del tratamiento de referencia, y esta diferencia fue estadísticamente significativa y se observó tan tempranamente como 4h después de la administración. De nuevo la diferencia parece clínicamente insignificante, no llegando a 4 puntos de 60 puntos que tiene la escala MADRS. Al analizar separadamente los resultados en función de que los sujetos declararan haber realizado al menos un intento previo de suicidio, la adición de esketamina nasal al tratamiento de referencia se mostró, de forma estadísticamente significativa, superior al tratamiento de referencia más placebo, pero de nuevo esta superioridad parece cuestionable clínicamente, al no llegar a 5 puntos MADRS, una reducción de 8 puntos MADRS equivalen a una mejoría mínima clínicamente observable [7]. Para sujetos sin intentos de suicidio previos no hubo diferencia estadísticamente significativa entre ambos. En el intervalo desde las 4h post-primera dosis y el día 25, fin del seguimiento, tanto los grupos con esketamina como con placebo siguieron mejorando, sin que la diferencia entre ambos aumentara al progresar el estudio. Ver Figura 7.
Las tasas de remisión (puntuación MADRS<12) fueron mayores en los grupos con esketamina que en los grupos placebo, ver Cuadro 6, esta diferencia no siempre alcanzó la significación estadística, aun sin hacer corrección por análisis múltiple, que tampoco se hace en ninguno de los contrastes planteados anteriormente. Además, la crítica de Moncrieff a estos puntos de corte es válida aquí también [8].
La herramienta para medir el riesgo de suicidio (Suicide Ideation and Behavior Assessment Tool) fue diseñada expresamente para los estudios ASPIRE [12, 13] por su sensibilidad al cambio, su principal componente es la escala de impresión clínica global de gravedad del riesgo de suicidio – revisada (CGI-SS-r), que toma valores entre 0 “normal y ausencia de riesgo de suicidio” y 6 “entre los pacientes más extremadamente suicidas”.
El riesgo de suicidio, tanto en los sujetos tratados con esketamina como en los tratados con placebo (ambos más el tratamiento de referencia), mejoró a las 24h de la primera dosis, medido con la escala CGI-SS-r, sin que hubiera diferencia estadísticamente significativa entre ambos grupos. La diferencia de medias entre los grupos fue -0,20 puntos (IC 95%: -0,43 a 0,04). Incluso la medición de la CGI-SS-r a las 4h postdosis rindió diferencias de cambio de dudoso significado clínico, -0,41 (IC 95%:-0,63 a -0,18) sobre una escala de 6 puntos. No se informa de valoraciones posteriores del riesgo de suicidio, por lo que el efecto de la esketamina sobre este, se desconoce más allá de las 24h [14].
Tanto la información sobre resultados de los estudios Tranform y Aspire que se incluyó en la ficha técnica [1], como el artículo publicado que informa del estudio Transform I [4] emplean el método de “observación basal arrastrada hacia delante” (BOCF) para el manejo de datos perdidos; consiste en que la valoración basal de un sujeto es también asignada como valoración final, si este abandona por cualquier razón del estudio, se basa en la asunción de que los sujetos que no finalizan un ensayo no se benefician del fármaco en estudio, y retornan al valor basal. Los informes publicados en revistas del estudio Aspire 2 [12], emplean el método de “última observación arrastrada” (LOCF), consiste en imputar el valor de la última observación disponible como valoración final; trata de reflejar el hecho de que los abandonos pueden producirse en diferentes momentos del estudio y los sujetos pueden haber experimentado algún grado de mejoría en el momento de salir del estudio, pero que no hay cambio entre la última medida y la salida, ni mejoran ni empeoran más.
En las publicaciones de los estudios Transform 2 [5] y 3 [6], Aspire 1 [11] y TRD2005 [9] se empleó el método denominado “modelo mixto de medidas repetidas” (MMRM), que utiliza una estimación basada en la probabilidad, efectos específicos del sujeto y correlaciones entre medidas repetidas, basándose en que las razones de abandono del estudio pueden estar o no relacionadas con el medicamento a investigación y que la distribución de las observaciones basales raramente es igual a la distribución de las respuestas de los pacientes en el estudio. Estos tres métodos están sujetos a un considerable riesgo de sesgo, por tanto se recomienda que al emplear uno de estos métodos se justifiquen las suposiciones sobre las que se basa su elección y además se emplee un método alternativo como análisis de sensibilidad para probar la robustez de los resultados [15]. En los distintos estudios presentados anteriormente, con diseños similares, se emplean diferentes métodos de análisis de los datos perdidos, en todos los casos se confirma la ausencia de relevancia clínica de los resultados.
La evaluación de seguridad a lo largo de los tres estudios TRANSFOM [4-6]consistió en el registro de los sucesos adversos emergentes durante el tratamiento y otras medidas de seguridad como hematología, química sérica, análisis de orina, examen físico, electrocardiograma, suicidabilidad (escala de puntuación de la gravedad de suicido de Columbia, C-SSRS), de cualquier solicitud de aumento de dosis o frecuencia de estas. Los signos vitales, escala puntuada por el clínico del estado disociativo (CADSS) y los 4 ítems sobre síntomas positivos de la escala breve de evaluación psiquiátrica (BPRS). Se evaluaron basalmente y luego antes de cada dosis y tras 40 minutos, 1 hora (solo signos vitales), y 1,5 horas después de cada dosis. La escala modificada de evaluación por el observador de la alerta/sedación (MOAA/S) (0, no responde a estímulos dolorosos a 5, responde a su nombre pronunciado en tono normal) cada 15 minutos, desde antes de cada dosis hasta 1,5 horas después.
Antes de abandonar la clínica de administración, los sujetos fueron evaluados con la evaluación clínica global de la preparación al alta 1 hora y 1,5 horas después de cada dosis (el momento más temprano para dar el alta) y cada 15 minutos posteriormente hasta que el sujeto estuviera en condiciones de ser dado de alta. La tolerabilidad nasal se evaluó con cuestionarios de síntomas y examen nasal directo a los sujetos de los estudios. Al finalizar el tratamiento se evaluó el potencial de abuso con la lista guía de retirada cumplimentada por el clínico (PWC-20) para evaluar los síntomas de retirada y una batería cognitiva (CogState®) para medir el potencial impacto de la esketamina en esta área. En el estudio SUSTAIN 1 la evaluación de seguridad siguió el mismo esquema [10], protocolo seguido también por un estudio abierto no controlado, SUSTAIN 2 [16]. La evaluación de seguridad en el estudio con población japonesa (TRD2005) fue similar, salvo que no aplico la batería de pruebas cognitivas [9]. En los estudios ASPIRE [12, 13] se registraron los eventos adversos a lo largo del estudio, y además se evaluaron los signos vitales, la CADSS y la MOAA/S en todas las visitas para administrar dosis.
Los informes de estos estudios solo recogen efectos adversos que se presentan en ≥5% de los sujetos participantes, lo cual deja fuera una importante fracción de efectos adversos que deben ser considerados como frecuentes al producirse con una frecuencia ≥1% y desde luego a todos los considerados como poco frecuentes (<1%). La notificación de efectos adversos en las publicaciones fue muy irregular y presenta algunas disparidades respecto a lo anotado en las bases de datos gubernamentales de registro de ensayos clínicos correspondientes. Al no ser informados claramente en las publicaciones de los ensayos clínicos, los datos sobre muertes, número de sujetos con efectos adversos graves y número de sujetos con al menos un efecto adverso los hemos tomados de la web “ClinicalTrials.gov” [17-24], ver Cuadro 7
Las reacciones adversas descritas con mayor frecuencia tras la administración de esketamina nasal fueron: mareos (31%), disociación (27%), náuseas (27%), cefaleas (23%), somnolencia (18%), disgeusia (18%), vértigo (16%), hipoestesia (11%), vómitos (11%) y aumento de la presión arterial (10%). El Cuadro 8 indica el tipo de reacciones adversos por esketamina y su frecuencia, recogidas en la ficha técnica de la AEM [1].
La disociación, se ha descrito en el 27% de los sujetos, se resuelve normalmente a los 90 min de la administración, fue grave en el 3-4% de ellos, su gravedad tendía a disminuir con la repetición de la administración. Otros términos relacionados fueron: desrealización (2,2%), despersonalización (2,2%), ilusiones (1,3%) y distorsión del tiempo (1,2%).
Sedación/somnolencia se presentaron respectivamente en el 9,3% y 18,2%, se resuelven normalmente a los 90 min tras administración, y su frecuencia fue relativamente estable en las administraciones repetidas.
Existe riesgo de deterioro neuropsiquiátrico y motor y de depresión respiratoria, tras la admin istración de esketamina se requiere la supervisión por un profesional sanitario y no se debe abandonar el centro de salud hasta que el paciente esté clínicamente estable.
Esketamina no ha mostrado prevenir el suicidio ni reducir las conductas o ideas suicidas; por lo que el riesgo persiste hasta que se logra una remisión significativa de los síntomas depresivos, el riesgo es mayor en las primeras fases de la recuperación. Esketamina debe emplearse con precaución en caso de antecedentes de psicosis o manía (riesgo de exacerbación). Los sujetos mayores de 65 años son más susceptibles a caídas y lesiones relacionadas con la sedación. El uso concomitante de esketamina con depresores del SNC (benzodiazepinas, opioides, alcohol y otros) aumentan los riesgos de sedación y depresión cardiorrespiratoria [1].
Cambios en la presión arterial, a los 40 min de la administración se producen aumentos de la presión sistólica de 7 a 9 mmHg, y en la diastólica de 2 a 5mmHg, y a los 90 min estos aumentos son solo de 2 a 5mmHg en la sistólica y de 1 a 3mmHg en la diastólica. Se produjo un aumento en la presión sistólica de >40mmHg en el 8% (<65 años) y en el 17% (≥65 años) y aumentos >25mmHg de la diastólica en el 13% (<65 años) y el 14% (≥65 años). La incidencia de urgencia hipertensiva por presión sistólica ≥180mmHg fue del 3% y por diastólica ≥110mmHg del 4%.
El aumento de la presión arterial es máximo alrededor de los 40min tras la administración y suele durar 1 a 2 h; si se mantiene elevada durante un tiempo más prolongado hay que solicitar asistencia médica para su control, si hubiera crisis hipertensiva (PA sistólica ≥18mmHg, o PA diastólica ≥120mmHg) deberá considerarse una urgencia médica. Es adecuado reducir la presión arterial antes de iniciar el tratamiento con esketamina.
Pacientes con enfermedades cardiovasculares significativas o inestables, requieren de administración en centros con medios y personal especializado en RCP. Entre las enfermedades que se deben tener en cuenta figuran, a título de ejemplo:
Se debe extremar la precaución al utilizar esketamina encasos de lesión cerebral, asi como con condiciones asociadas a hipertensión intracraneal, incluyendo la terapia intratecal (riesgo de daño cerebral), y casos de hipertiroidismo no tratado (riesgo de hipertensión). La asociación con psicoestimulantes (anfetaminas y relacionados) y medicamentos hipertensores (antidepresivos IMO, ergometrina, vasopresina, hormonas tiroides) aumentan el riesgo de aumento de la presión arterial y/o intracraneal [1].
Deterioro cognitivo y de la memoria. En los ensayos clínicos el rendimiento cognitivo, medido con la batería CogState®, se mantuvo estable. Aunque solo los estudios Tranform 3 [6] y Sustain 2 [16] informan al respecto, indicando respectivamente que no hay diferencias en las medidas cognitivas entre el grupo con esketamina y el grupo placebo, ni al progresar el tratamiento. Los resultados sobre las variables cognitivas en los estudios Transform 1 y 2 [4.5] y Sustain 1 [10] no se indican en los informes respectivos, en los que solo se informa de que serán objeto de una publicación separada, que al parecer en el momento de redactar este artículo no ha visto la luz.
Un ensayo clínico [25] controlado con placebo, cruzado, de fase 1, mostró que una dosis aislada de esketamina nasal empeora el rendimiento cognitivo, medido con la batería CogState®, a los 40 minutos postdosis, pero este efecto se había desvanecido al medirlo a las 2, 4 o 6 horas postdosis, pero la situación puede ser completamente distinta con el uso reiterado a largo plazo. Con el uso continuado y frecuente de ketamina se han detectado deterioros en estas áreas [26].
La escala CogState o CBB (CogState Brief Battery) [27] fue creada por la compañía CogState Ltd (ASX: CGS) proveedor líder de soluciones de ciencia y tecnología dedicado a optimizar la medición de la cognición en ensayos clínicos, la investigación académica y la atención médica. Entre sus principales clientes figuran las principales compañías biofarmacéuticas del mundo, organizaciones deportivas de élite y militares, instituciones académicas de renombre y alianzas público-privadas. La escala se diseñó con el objetivo en crear tareas trans-culturales válidas que requiriesen mínimas instrucciones y respuestas verbales. Incorpora diferentes tareas que evalúan tiempo de reacción (simple y de elección), memoria de trabajo y verbal, aprendizaje visual y verbal, función ejecutiva y aprendizaje visual asociado y según sus autores, puede detectar cambios sutiles con el uso y abuso de las drogas. Sin embargo, no se tomaron en cuenta que las funciones de atención y memoria de trabajo son dos funciones independientes, pero íntimamente relacionadas, y siempre se deben evaluar por separado, aplicando otras pruebas que confirmen los datos encontrados.
Es necesario realizar en un mismo contexto otras pruebas neuropsicológicas, que permitan descartar otros déficits cognitivos causados por el fármaco. Por ello, una evaluación neurocognitiva eficaz no solo debe evaluar el proceso de aprendizaje sino también la memoria a largo plazo, como mínimo dos a tres semanas después de terminado el ensayo clínico, cuando la información tanto visual, auditiva y verbal ya está consolidada en la neocorteza.
Síntomas del tracto urinario. Se ha comunicado cistitis intersticial con el uso nasal de ketamina [28]. El padecimiento de depresión y el uso prolongado de ketamina también se han asociado con un aumento del riesgo de trastornos urinarios [29]. En los ensayos clínicos con esketamina nasal se ha observado poliaquiuria, disuria, urgencia de micción, nicturia y cistitis [1].
Abuso de drogas, dependencia y abstinencia. Con el uso prolongado de ketamina se ha descrito dependencia y tolerancia, al interrumpir el suministro se ha descrito búsqueda compulsiva de la sustancia, ansiedad, temblores, sudoración y palpitaciones. La ketamina ha sido objeto de usos desviados y abusivos. El uso bajo la supervisión de un profesional sanitario realmente no evitaría el uso desviado de esketamina. Ketamina es un medicamento de manejo por el médico anestesista o un médico intensivista en hospitales o por el veterinario, lo que no ha evitado que se convierta en sustancia de abuso.
Además, el paciente dependiente de esketamina podría acudir a la búsqueda de ketamina. En policonsumidores (n=41) dosis nasales de esketamina (84 y 112mg) o de ketamina intravenosa (0,5mg/kg en 40 min) como control activo, rindieron puntuaciones subjetivas de apetencia de la sustancia, indicadoras del riesgo de abuso, significativamente mayores que placebo [1]. Tras el tratamiento con esketamina nasal, el fabricante indica que no se requiere reducción gradual de la dosis, dice que el riesgo de síndrome de abstinencia es bajo, según los datos de los ensayos clínicos [1]. Pero el ensayo Sustain 1[10, 21] fue de retirada y puede dar algo de información de los riesgos que esta acción conlleva, aun así, la duración previa a la retirada de uso de esketamina en este estudio es relativamente pequeña, por lo que se debe ser precavido, especialmente si se considera que si se ha descrito síndrome de abstinencia a ketamina [30, 31]. La situación extraordinariamente controlada que implica la realización de un ensayo clínico, dista astralmente de la situación que se produce una vez comercializado el producto.
Un estudio reciente [32] publicado dos años después de la aprobación de esketamina nasal, identificó señales de alerta de seguridad relacionadas con este medicamento, e indicó que se debe prestar más atención a un conjunto de acontecimientos adversos: trastornos sensoriales, amnesia, trastorno cognitivo, síndrome serotonérgico, akatisia, hipocinesia y parálisis. Estos efectos no se detectaron previamente en los ensayos clínicos precomercialización, y solo se han informado en las bases de datos de farmacovigilancia. Las publicaciones de los ensayos clínicos están firmadas por autores con extensos conflictos de interés [4-6, 9-10, 12-14, 16, 25].
Datos farmacocinéticos de esketamina [1]. La biodisponibilidad de una dosis de 84mg de esketamina administrada en pulverización nasal es aproxi5adamente 48%. La absorción es rápida, pudiendo detectarse en sangre a los 7 minutos y alcanzando la concentración máxima entre los 20 y 40 minutos. En el rango de dosis de 28 a 84mg, los aumentos de Cpmax y AUC∞ fueron menos que proporcionales a los aumentos de dosis de 28mg a 56mg u 84mg, pero casi proporcionales de 56mg a 84mg. El 44% de la concentración plasmática se encuentra fijada a proteínas, cantidad que es independiente de la función hepática. El volumen de distribución en condiciones de equilibrio (cuando se administra por via IV) es de 709 litros.
La esketamina no es sustrato de la glucoproteina P (Pgp) de multirresistencia a fármacos, ni de la proteína de resistencia al cáncer de mama (BCRP), ni del transportador de aniones orgánicos (OATP1B1 y OATP1B3). Tampoco inhibe a este transportador, ni al transportador de cationes orgánicos (OCTP1, OCTP2 y OCTP3), ni al transportador de expulsión de múltiples fármacos y tóxicos (MATE1 y MATE2). Se metaboliza ampliamente en el hígado, principalmente por N-desmetilación formándose noresketamina, con la participación los CYP2B6 y CYP3A4 y en menor proporción los CYP2C19 y CYP2C9.
La noresketamina se metaboliza posteriormente con participación del sistema CYP, produciéndose otros metabolitos que posteriormente sufren glucurocongugación. El aclaramiento plasmático (tras administración iv) es de unos 89 litros/hora. La eliminación es mayoritariamente por la orina (80%), fundamentalmente en forma ya metabolizada, <1% como esketamina inalterada; solo un 2% se elimina por las heces (incluso tras la administración oral). La semivida terminal de eliminación tras administración nasal oscila entre 7h y 12h, lo que implica la posibilidad de persistencia de efectos adversos residuales durante varias horas tras la administración, como disminución de reflejos y dificultad de manejo de maquinaria peligrosa. El perfil farmacocinético tras la administración de dosis múltiples dos veces por semana es similar al de la administración de dosis única, ya que con este esquema no se produce acumulación, lo que si ocurriría si se administraran dosis más frecuentemente.
Interacciones farmacocinéticas [1] . No parece que los inductores ni los inhibidores de los enzimas hepáticos interfieran de forma clínicamente significativa con la farmacocinética de la esketamina, ni que esta interfiera sobre la de otros medicamentos. Ver Cuadro 9.
Efecto de determinadas condiciones sobre la farmacocinética [1]: La edad aumenta Cpmax y AUC∞,ver Cuadro 10. Tras la administración nasal de dosis de 28mg de esketamina, la insuficiencia renal y la insuficiencia hepática aumentan Cpmax y AUC∞. No se ha estudiado el efecto de la diálisis, ni de la insuficiencia hepática grave, ver Cuadro 11. Los sujetos asiáticos presentan un Telim1/2 superior que los caucásicos (7,1-8,8h versus 6,8h), al igual que AUC∞, pero el efecto sobre la Cpmax dependió del subgrupo estudiado, ver Cuadro 12. Ni el sexo, ni el peso (rango estudiado de >39Kg a 170kg), ni la rinitis alérgica parecen ejercer influencia sobre la farmacocinética. No se precisa ajuste de dosis en caso de insuficiencia renal, incluso grave, pero se carece de estudios en casos de diálisis. En sujetos con insuficiencia hepática moderada (clase B Child-Plugh) se debe emplear con precaución la dosis de esketamina de 84mg. No se recomienda emplearla si es grave (clase C Child-Plugh). El uso continuado de ketamina se ha asociado a hepatotoxicidad y a síntomas del tracto urinario (infección del tracto urinario conocida como cistitis ulcerosa).
No se recomienda el empleo de esketamina en mujeres embarazada, nodrizas, ni en aquellas en edad fértil que no estén recibiendo un método anticonceptivo. La experimentación animal ha mostrado que ketamina produce neurotoxicidad en los fetos en desarrollo y que pasa a la leche materna. Ketamina no ha mostrado afectar negativamente la capacidad reproductora en animales. Cuando se administran a animales dosis que causan anestesia ligera durante periodos de crecimiento cerebral rápido o de sinaptogénesis, originan pérdida celular en el cerebro en desarrollo que puede asociarse a dificultades cognitivas prolongadas. Cuando se administraron a conejos dosis que causaban toxicidad a las madres, se observaron malformaciones esqueléticas y reducciones de peso en los embriones. Se han observado efectos genotóxicos en una prueba de micronúcleos in vitro en presencia de actividad metabólica [1].
Población pediátrica. No se recomienda en menores de 18 años. Aunque se han realizado estudios en grupos de menores con trastorno depresivo mayor, no se han presentado los resultados de los mismos (a la fecha de la redacción de estas notas el plazo concedido para su publicación había expirado, por lo que fue preciso solicitar una prórroga para su comunicación a la Agencia Europea del Medicamento, que se ha concedido). [1]
Proceso de prescripción y administración [1]. El medicamento debe ser prescrito por un psiquiatra, y su administración la realizará el propio paciente que debe ser supervisada directamente por un “profesional sanitario”, que también le observará durante “un periodo post-administración y en un entorno clínico adecuados”. Esketamina nasal no se debe emplear en caso de hipersensibilidad a esta sustancia o a alguno de los excipientes que se añaden al preparado farmacéutico (ácido cítrico, edetato sódico, hidróxido sódico y agua) y ni en aquellas situaciones en las que un aumento de la presión arterial o la presión intracraneal suponga un riesgo grave (valvulopatías aneurismáticas, antecedentes de hemorragia intracerebral, y episodio caridovascular en las 6 semanas previas).
Antes del tratamiento [1] se debe medir la presión arterial del sujeto, y “si está elevada”, reconsiderar el beneficio riesgo. No se debe administrar si un aumento de esta o de la presión intracraneal supone un riesgo grave para el paciente. Si el sujeto padece enfermedad cardiovascular o respiratoria previa clínicamente significativa o inestable, se debe administrar en un entorno que disponga de equipo y profesionales adecuados para la reanimación cardiopulmonar. Se aconseja que, para evitar náuseas y vómitos, no se ingieran alimentos sólidos ni líquidos desde, respectivamente, 2h y 30min antes de la administración de esketamina. Tampoco se deben administrara otros medicamentos por vía nasal como descongestionantes y corticoides en la hora previa.
El esquema posológico [1] es diferente según la indicación y la edad del sujeto.
Trastorno depresivo mayor resistente al tratamiento (TDMR), se distinguen 2 fases, de inducción (semanas 1 a 4) y de mantenimiento (desde la semana 5º). En adultos menores de 65 años. La dosis inicial será de 56mg, y las siguientes (hasta semana 4º inclusive) de 56 a 84mg, dos veces por semana, en función de la eficacia y tolerabilidad de la dosis previa. Al finalizar la semana 4 se debe evaluar los efectos beneficiosos a fin de considerar si se debe continuar el tratamiento. En las semanas 5º a 8º se administrará una vez por semana, a la misma dosis empleada al final de la fase de inducción. A partir de la semana 9º y las siguientes, las dosis se administrarán cada 1 o 2 semanas, debiéndose emplear con la menor frecuencia posible para mantener la remisión/respuesta. La necesidad de continuar el tratamiento “se reexaminará periódicamente”. Una vez mejorados los síntomas se recomienda continuar el tratamiento durante al menos 6 meses. En mayores de 65 años, el plan posológico es idéntico, con la excepción de que la dosis inicial será de 28mg, y las subsiguientes podrán ser entre 28 y 84mg. No se ha estudiado en menores de 18 años.
Tratamiento agudo a corto plazo de la emergencia psiquiátrica debida a trastorno depresivo mayor, en adultos menores de 65 años, durante 4 semanas se administrará una dosis de 84mg dos veces por semana, reduciendo a 56mg según tolerabilidad. Después de este periodo la medicación antidepresiva oral debe continuar a criterio clínico. En estos pacientes el tratamiento con esketamina debe formar parte de un tratamiento integral. No se ha estudiado para esta indicación en mayores de 65 años, ni en menores de 18 años. Si bien el suicidio es un importantísimo problema de salud pública, la propia ficha técnica de la sustancia reconoce que la esketamina nunca ha probado reducir la incidencia de suicidios.
El dispositivo [1] para administración nasal dispensa dos pulverizaciones con dosis de esketamina de 14mg, una por cada fosa nasal (total 28mg por dispositivo). No se debe preparar (cebar) el dispositivo hasta justo antes de su uso, para evitar pérdida de principio activo. Para la dosis de 56mg se precisan dos dispositivos, y tres dispositivos para la de 84mg. Ha de descansarse un tiempo de 5 minutos entre el uso de 2 dispositivos. Ver Figuras 8 y 9
Si se omiten sesiones durante las primeras 4 semanas, los pacientes deben continuar con su posología actual. Si se omiten sesiones de mantenimiento y se produce empeoramiento de los síntomas se considerará a juicio clínico volver a la pauta posológica anterior. No debe emplearse un dispositivo de sustitución en caso de estornudo inmediatamente después de la administración, ni tras la administración de dos pulverizaciones consecutivas en la misma fosa nasal [1].

Tras la administración[1] se debe volver a medir la presión arterial a los 40 minutos y posteriormente “si está clínicamente justificado”. Debido a la posibilidad de sedación, disociación y elevación de la presión arterial, el paciente debe ser vigilado por un profesional sanitario hasta que esté clínicamente estable y listo para abandonar el centro sanitario, usualmente a los 90 a 120 minutos de la administración. Básicamente, comprobar que la tensión arterial del sujeto está normalizada, que el estado de alerta es adecuado y que no presenta síntomas de disociación psíquica, así como que se le ha informado que no debe manejar vehículos y la conveniencia de descansar hasta el día siguiente. Esketamina pude causar sedación, disociación, alteración de la percepción, mareos, vértigos y ansiedad, que incapacitan para el manejo de vehículos. Los usuarios de esketamina deben abstenerse de conducir vehículos y manejar máquinas hasta el día siguiente después de un sueño reparador. A continuación, se muestra la lista guía de comprobación previa al abandono del centro sanitario tras la administración de esketamina (ver Cuadro 13).
El precio propuesto por la empresa farmacéutica es de €522 a €783 por sesión de tratamiento, según la dosis que necesite el paciente [33], a ello habría que añadir el coste las instalaciones y personal especializado necesarios para la administración. En España el medicamento no ha logrado ser financiado por el sistema de salud y permanece sin comercializar [34, 35]. En Argentina el precio es de 46.000 pesos la dosis de 56mg y de 69.000 pesos la de 84mg [36]. En USA el precio es de US$140 a US$240 [37]. En UK el precio por curso de tratamiento con esketamina es del orden de £10.000 Libras [38]. Mejorar un trastorno depresivo grave resistente al tratamiento o una muerte por suicidio tiene un extraordinario valor, que puede reflejarse en el precio del medicamento. El problema es que en el caso de esketamina nasal estas ventajas son eslóganes comerciales más que realidades objetivables.
Un poco de historia. Esketamina pertenece la familia de los anestésicos arilciclohexilaminas, también conocido como anestésicos disociativos. El grupo se inauguró con el descubrimiento de 1926 de la fenciclidina (“polvo de ángel” o PCP), pero no fue hasta la década de los 1950 cuando comenzó a usarse como anestésico en clínica humana, aunque pronto, en la década de los 1960 se detectó el uso como sustancia de abuso y se abandonó el uso clínico humano, quedando relegada como anestésico solo de uso veterinario. En la década de los 1970 el abuso de fenciclidina alcanzó proporciones epidémicas entre los jóvenes de los países desarrollados, lo que dio lugar a su inclusión en las listas de sustancias controladas, a pesar de ello el abuso aún persiste. La ketamina es un derivado de la fenciclidina que se introdujo como anestésico en clínica humana en la década de los 1970; siendo más manejable que fenciclidina al poseer una menor potencia y una eliminación más rápida.
El uso abusivo de ketamina también se hizo pronto evidente, no parando de aumentar hasta la actualidad, tanto por sus propiedades disociativa como por su capacidad de inducir sumisión química [39]. La psiquiatría siempre había mostrado interés por el uso terapéutico de las sustancias psicodislépticas [40]. En la década de los 2000 se sugirió que ketamina pudiera tener, tras dosis única intravenosa, un efecto positivo sobre la depresión y las ideas suicidas (no sobre los actos suicidas), que aunque rápido era de corta duración y requería varias dosis semanales, además surgieron dudas acerca del mantenimiento de su efecto tras las 3 semanas de uso [41]. Para solventar estos problemas se desarrolló el preparado con esketamina nasal, pero como hemos analizado anteriormente, el producto dista de ser medianamente satisfactorio para el fin que se propone. Ahora los ojos se vuelven hacia el otro enantiómero, conocido como arketamina, al que se le suponen propiedades antidepresivas más potentes y duraderas que las de la esketamina y con menos problemas conductuales y de abuso, basándose en estudios en animales, pero solo se dispone de un estudio abierto no controlado con esta sustancia, en dosis única administrada a siete sujetos con depresión resistente, llevado a cabo por autores con notables conflictos de interés [42].
Conclusión. Esketamina nasal no parece mejorar de manera apreciable la depresión resistente al tratamiento, ni parece aliviar tampoco de manera significativa los riesgos asociados a urgencia psiquiátrica relacionada con un trastorno depresivo mayor, a esta falta de eficacia se asocia un riego sustancial de efectos adversos evidentes y peligrosos. El fabricante hace referencia a un bajo riesgo de abuso, pero es una afirmación gratuita, esto junto con los potenciales efectos cognitivos a largo plazo son motivos de precaución. Una vez más el uso de enantiómeros se aprovecha para obtener nuevas patentes e incrementar el precio de medicamentos antiguos.
Mejorar un trastorno depresivo grave resistente al tratamiento o una muerte por suicidio tiene un extraordinario valor, pero el que comercializa la esketamina nasal utiliza este dolor para obtener pingües beneficios con un medicamento inútil e inseguro. Considerando la falta de eficacia y las dudas de seguridad, cuanto menos resulta sorprendente la aprobación de la comercialización por organismos como la FDA y la AEM de la esketamina nasal como tratamiento para la depresión resistente y las urgencias psiquiátricas por episodio depresivo.
Sobre los autores
Marín S. Licenciada en Psicología, Máster en neuropsicología, Doctora en psicología del desarrollo. Psicólogo habilitada en clínica en el Hospital de día del Centro Dr. Esquerdo.
Martínez F. Licenciado en Farmacia, Especialista en Farmacia hospitalaria, Máster en Salud mental y drogodependencia, Profesor asistencial asociado UMH. Doctorando en Ciencias de la salud UA. Jefe de Unidad de Farmacia del Centro Dr. Esquerdo.
Ortiz R. Graduado en Farmacia, Máster en administración y Gestión de Oficinas de Farmacia, Doctorando en Toxicología UMH.
Pellín C. Licenciada en Farmacia, Doctora en Toxicología, Profesora titular de Toxicología UMH.
Pol E. Licenciado en Farmacia, Especialista en Farmacia hospitalaria, Máster en Medicina humanitaria, Doctor en Medicina experimental.
Bibliografía