Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
Investigaciones
La descoordinación entre países para autorizar medicamentos
Nalili Lepetit-Chella
Fernando Anido
El Mundo, 17 de enero de 2016
http://www.elmundo.es/grafico/salud/2017/01/16/57c9a48222601d5a698b458b.html
La Agencia Europea de Medicamentos puede tomar decisiones centralizadas, pero no existe un sistema a nivel mundial.
Incluso en la Unión Europea, medicamentos retirados del mercado por algunos países están a la venta en otros (Ver http://www.elmundo.es/salud/2017/01/16/57e8e622e5fdea707f8b4598.html)
Descubra los diez casos de esta investigación con una aplicación (Ver http://www.elmundo.es/grafico/salud/2017/01/16/57c99d8c22601d187d8b458c.html)
Efectos secundarios del mercado de la salud (Ver http://www.elmundo.es/salud/2017/01/16/57cd851622601d2a368b45f3.html)
“Este medicamento me ha echado a perder. Sí, es eso: el Agreal me ha echado a perder.” Anne-Marie era secretaria médica en Francia cuando tomó este fármaco contra los sofocos de la menopausia. Tenía 52 años, y su ginecóloga le aconsejó el Agreal. Ahí comenzaron sus problemas de salud. Era mayo de 2002. Primero fue la vista, que a veces se le ponía borrosa. Luego tuvo tortícolis repetidas veces y sus uñas se desdoblaron. Después empezaron los temblores. No entendía qué le pasaba, pero lo atribuyó al estrés y al cansancio por el trabajo.
En 2004, tras una pausa de unos días en el tratamiento, volvió a tomar Agreal. Los temblores regresaron el mismo día, más fuertes que nunca. Se preguntó si era por tomar el fármaco. Cogió el prospecto y lo leyó: como efectos secundarios aparecían los síntomas del Parkinson. Anne-Marie paró inmediatamente el tratamiento, con el visto bueno de su ginecóloga. Tres años después, la Unión Europea prohibió el Agreal. Fue entonces cuando descubrió que el principio activo del medicamento, la veraliprida, era un neuroléptico: un tipo de medicamento generalmente indicado en casos de psicosis. España lo había retirado desde 2005 por sus efectos adversos neurológicos y psiquiátricos.
Este desfase entre países no es una excepción. Y aunque la Unión Europea ha empezado a armonizar la retirada de medicamentos peligrosos, tal sistema no existe a escala mundial. Un Estado puede considerar que una sustancia conlleva más riesgos que beneficios para los pacientes, pero el país vecino no tiene por qué compartir su decisión. Las agencias de medicamentos nacionales mantienen una comunicación entre ellas, pero toman decisiones de forma individual. Mientras las multinacionales farmacéuticas juegan en un terreno global, la farmacovigilancia, el sistema de notificación de efectos adversos, está limitada por las fronteras nacionales.
Tras la recogida de datos en los registros de 38 países, se han localizado diez sustancias activas que ponen en evidencia la falta de coordinación global en el control de medicamentos. Medicamentos prohibidos en algunos Estados por ser peligrosos, y a veces poco eficaces, pero que siguen siendo comercializados durante años en otros. Algunos están a la venta en la actualidad. Germán Velásquez, economista colombiano que trabajó durante 20 años para la Organización Mundial de la Salud (OMS), lo afirma sin vacilar: “El mercado mundial de medicamentos está totalmente regido por una lógica comercial”.
Anne-Marie no pudo aplicar esa lógica comercial. A principios de 2005 abandonó su puesto en el consultorio médico. Tenía 55 años y nunca volvió a encontrar trabajo. Seis meses después de dejar de tomar Agreal, su salud había empeorado. Los temblores persistían por periodos, le resultaba cada vez más difícil concentrarse y, sobre todo, se hundía en una depresión. “Pensaba que no podía asumir mi carga de trabajo, que era demasiado estrés. Que podía ser la menopausia”, recuerda la jubilada. Así, después de trabajar durante las semanas de Navidad y de Año Nuevo en lugar de tener vacaciones, no pudo más y dejó su empleo: “Me derrumbé”.
Tardó casi tres años en entender qué le pasaba. Fue un día de diciembre, cuando el diario regional francés La Dépêche du Midi publicó un artículo titulado “Medicamentos: demasiados accidentes” (http://www.ladepeche.fr/article/2007/12/14/419126-sante-medicaments-trop-d-accidents.html ). Y allí estaba: el Agreal. El texto explicaba que su retirada del mercado europeo, confirmada por la Comisión Europea dos meses antes, era consecuencia de la “sobrevenida de diferentes efectos secundarios neurológicos y psiquiátricos”. Anne-Marie sintió alivio en ese momento: “Entendí que no era yo la que se estaba volviendo loca”. Pero al poco tiempo, la noticia adquirió matices amargos: “Si hubiera entendido que era el Agreal, si lo hubiera sabido, nunca hubiera dimitido. He perdido diez años de cotización para mi jubilación”.
La veraliprida ha sido retirada de la mayor parte del mundo. Pero sigue siendo comercializada en México bajo el nombre de Aclimafel.
“Eso pasa con mucha frecuencia”, sostiene Germán Velásquez, refiriéndose a casos similares al de la veraliprida: casos en los que unos países prohíben un medicamento por considerarlo más peligroso que saludable, mientras que otros lo dejan a la venta. Esta investigación ha profundizado en diez principios activos problemáticos, incluida la veraliprida. Entre ellos, un medicamento que dio mucho que hablar en Francia: el Mediator, comercializado por los laboratorios franceses Servier. Destinado para ayudar a adelgazar a pacientes con diabetes, se recetó también a personas sin diabetes. Un estudio de la doctora Irène Frachon (https://www.editions-dialogues.fr/livre/mediator-150-mg/) mostró el vínculo entre el principio activo del Mediator, el benfluorex, y problemas cardiacos y vasculares: podía provocar una enfermedad del corazón llamada valvulopatía cardiaca e hipertensión de las arterias pulmonares.
La Comisión Europea prohibió esa sustancia en junio de 2010, tras una recomendación de la Agencia Europea de Medicamentos (EMA). Pero 13 años antes, la agencia suiza de regulación de medicamentos, Swissmedic, ya se había preocupado por la seguridad del benfluorex y los laboratorios Servier lo habían retirado de ese país en 1998. Cinco años más tarde, España prohibió el Modulator, nombre de marca del benfluorex en el país, por su posible implicación en valvulopatías cardiacas. Pero el Mediator siguió a la venta en Francia y Portugal hasta noviembre de 2009, causando a largo plazo entre 1.300 y 1.800 víctimas mortales galas, según un informe de la Fiscalía de París de 2013 (http://www.lemonde.fr/sante/article/2013/04/12/mediator-entre-1-300-et-1-800-morts-causees-a-long-terme_3158818_1651302.html ).
En el caso del dextropropoxifeno, un principio activo contra el dolor, un estudio publicado en 2004 le imputó 200 muertes al año entre 1992 y 1999, sólo en Suecia. La sustancia fue comercializada allí desde 1966 hasta 2011. Birgitta Jonasson, doctora en Medicina Forense, y Ulf Jonasson, doctor en Salud Pública, son los autores del estudio. Ambos luchan desde principios de los años 2000 por la retirada de esta sustancia del mercado mundial de medicamentos dado el número de muertes que le atribuyen. En 2010, la EMA estableció en un informe sobre la reevaluación del principio activo (http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/dextropropoxyphene_31/WC500014076.pdf ) que el mayor problema con esta sustancia era que “los pacientes podrían fácilmente tomar demasiado dextropropoxifeno y arriesgarse a una sobredosis mortal”.
Suiza fue el primer país que tomó medidas: prohibió en 2003 la última medicina con dextropropoxifeno que estaba autorizada en el país, el Distalgesic. Seis años después, la agencia europea, cuya sede está en Londres, recomendó lo mismo y fue imitada: varios países fuera de la UE siguieron su consejo. Por su parte, Estados Unidos, Canadá y Brasil negociaron con las farmacéuticas para que dejaran de comercializarlo dentro de sus fronteras. Pese a este movimiento, Argentina, Australia y China todavía autorizan la venta de medicinas con dextropropoxifeno, según consta en sus registros nacionales de medicamentos.
Organización Mundial de la Salud: ¿la solución?
En su web, la OMS se presenta como la autoridad directiva y coordinadora de la acción sanitaria en el sistema de las Naciones Unidas. Entre sus principales ámbitos de actividad aparecen los “sistemas de salud”. Pero su labor dirigente no llega a estos sistemas, en los que las decisiones individuales de cada país determinan la norma.
Para Germán Velásquez, que ahora trabaja en la organización intergubernamental de países en desarrollo South Center, la OMS debería crear una “red de información para todos los países en la que, en el momento en que uno de ellos encuentre un problema grave a un medicamento, avise al resto para que puedan estudiar si lo retiran o no”.
El primer paso para ello ya se dio. En abril de 2015, la OMS lanzó VigiAccess (http://www.vigiaccess.org/), una aplicación que da acceso a VigiBase: su base de datos mundial de las reacciones adversas (ADR por sus siglas en inglés) que se sospecha que han causado unos 100.000 productos medicinales. Pero cuando se busca, por ejemplo, el benfluorex (cuyo nombre de marca era Modulator en España), sólo aparecen siete casos de resultados mortales: una cifra muy alejada de las estimaciones de la Fiscalía de París, que considera que esta sustancia provocó entre 1.300 y 1.800 muertes en Francia.
El Centro de Monitorización de Uppsala (Suecia), que gestiona VigiBase desde 1978, advierte al público de que la base de datos “no debe ser considerada como un registro sistemático de los efectos secundarios que ocurrieron o que puedan ocurrir”. Más abajo, subraya otra de las limitaciones de VigiAccess: no informa sobre el número de pacientes que han sido expuestos a los productos, “así que (…) no es posible calcular la frecuencia de cualquier ADR”.
Infranotificación, el problema de la farmacovigilancia
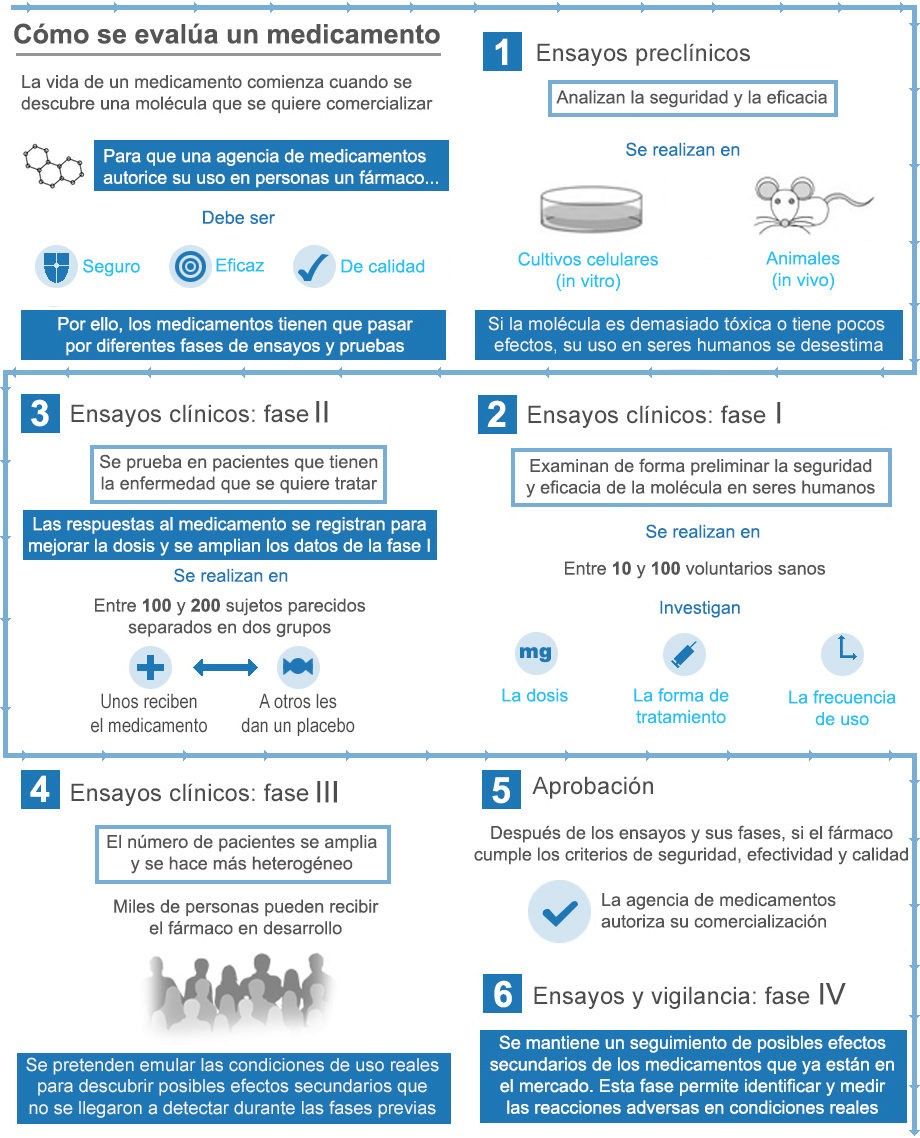
Los registros de reacciones adversas, ya sean de la OMS o de los Estados, sirven para controlar los efectos de un medicamento después de su comercialización. Primero, se realizan ensayos preclínicos -sobre animales- y clínicos -sobre centenares de personas-, pero éstos pueden no desvelar problemas que surgirán cuando la sustancia sea distribuida a gran escala.
La farmacovigilancia, que se basa en esos registros, desempeña un papel principal para regular las medicinas. Pero la complejidad de este sistema radica en que se parte de la base de que pueda existir una relación directa entre la toma de un medicamento y unos síntomas de origen desconocido. El posible vínculo de causalidad no es evidente cuando los trastornos que se aprecian no forman parte de los efectos secundarios conocidos de la sustancia. En un informe de 2012 sobre la “Vigilancia de la seguridad de medicamentos” (http://apps.who.int/medicinedocs/documents/s21836es/s21836es.pdf), la OMS reconoce que “la infranotificación de reacciones adversas a los medicamentos es un problema”.
La importancia de este fenómeno se puso de relieve en un artículo científico (https://people.eecs.berkeley.edu/~daw/teaching/c79-s13/readings/AdverseDrugReactions.pdf) de 2006 escrito por dos miembros de la Unidad de Investigación de la Seguridad de Medicamentos de Southampton (Reino Unido), Lorna Hazell y Saad Shakir. Al recopilar todos los estudios sobre el tema en aquel momento establecieron que, de media, se notificaban un 6% de las reacciones adversas. Ocho años después, la Universidad brasileña Estadual Paulista publicó un trabajo (http://www.scielo.br/pdf/reeusp/v48n4/es_0080-6234-reeusp-48-04-739.pdf) sobre el mismo asunto en el que llegó a una conclusión similar: estimaron que “entre el 5% y el 10% de las Reacciones Adversas a Medicamentos [eran] notificadas”.
Para remediar esto, la EMA implementó una nueva legislación en julio de 2012 ( http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/general/general_content_000491.jsp) que permite que, además de los médicos y de los farmacéuticos, los pacientes puedan comunicar sus propias reacciones adversas a las agencias de regulación en toda Europa. Pero para el Centro Belga de Información Farmacéutica (CBIP), una asociación de profesionales de la salud sin ánimo de lucro, el cambio no fue muy perceptible. “Pocos pacientes saben que lo pueden hacer, al menos en Bélgica”, afirma el doctor Thierry Christiaens, redactor jefe del CBIP y profesor de farmacología en la Universidad de Gante. En España, según la agencia de medicamentos (AEMPS), “las notificaciones de los ciudadanos corresponden a cerca de 1.000 de las 17.000 notificaciones anuales de sospechas de reacciones adversas a medicamentos recibidas”. Es decir, a menos del 6% de ellas.
Barreras de protección sueltas
En España, un laboratorio u organismo ‘promotor’ que desarrolla un medicamento tiene que seguir varios pasos antes de poder comercializarlo. El objetivo es probar ante una agencia de regulación de fármacos que la medicina es segura, eficaz y de calidad para un tipo de población determinado. Para ello, la institución se basa en los resultados de varios ensayos proporcionados por el ‘promotor’ del fármaco.
Estas pruebas las realizan técnicos contratados por el laboratorio. “Estimo que no sean parciales”, explica el catedrático de la Universidad Complutense Antonio González Bueno, “pero quien paga a esos técnicos es el laboratorio que quiere comercializar el producto”.
Para limitar estos riesgos de parcialidad, la Ley española prevé, de acuerdo con las directivas europeas, que esos ensayos deben ser supervisados por órganos “independientes”: los Comités de Ética de la Investigación Clínica (CEICs). Estos grupos de profesionales sanitarios y no sanitarios se encargan de vigilar que se respete la conformidad del ensayo a las normas legales así como la seguridad y los derechos de las personas que participan en ellos.
Al final, los CEICs emiten un dictamen para aprobar o no el ensayo. Si el fallo es positivo, el ‘promotor’ podrá someter los resultados de su ensayo a la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) o a la Agencia Europea de Medicamentos (EMA). Después, el organismo de regulación determinará si autoriza o no la comercialización del fármaco
La independencia de los CEICs es fundamental. En ningún caso pueden recibir una remuneración, sea directa o indirecta, de los promotores, por ejemplo. Sin embargo, a veces varios CEICs se encargan de evaluar un mismo ensayo. En estos casos, uno sirve de referencia a los otros y sólo su aprobación es necesaria para que sea considerado válido. Y aquí se rompe la cadena de independencia: es el promotor el que elige qué grupo llevará la voz cantante, según las instrucciones de la propia AEMPS.
Existen Comités de ensayos clínicos desde 1978 en España. Su papel y funcionamiento fueron consolidados y armonizados con toda la Unión Europea a partir del Real Decreto 223/2004. “Eso se ha reforzado muchísimo, pero los antiguos medicamentos, hay que ver cómo se hacían”, cuenta María Teresa Alfonso Galán, profesora de ciencias médicas de la Universidad de Alcalá, remitiéndose a los años setenta. Y se apoya en el caso de la veraliprida: “Hoy no pasaría un Comité de Ética de la Investigación. No tiene sentido que para los sofocos de la menopausia, que no son una enfermedad, se dé un neuroléptico: un medicamento contra la esquizofrenia”. Una mañana a finales de abril de este año, la doctora, con su colega Antonio Piga, profesor emérito, explicaban en una entrevista personal lo que saben de esta sustancia. Ambos han trabajado juntos durante más de 10 años para demostrar que el prospecto original del Agreal era incompleto.
El buflomedil también fue aprobado por primera vez en los años setenta. Está indicado para tratar la claudicación intermitente: dolores en las piernas que causan problemas para andar. La cardióloga Tine de Backer, de la Universidad de Gante (Bélgica), considera que es un buen ejemplo de los beneficios que tendría la reevaluación de los productos antiguos. Critica que muchos medicamentos antiguos “pueden no haber sido sometidos a un análisis formal de beneficios-riesgos”.
Un problema que, para el Centro Belga de Información Farmacéutica, podría tener una solución: proponen instaurar “un registro periódico de todos los medicamentos cada tres a cinco años”, y evaluar cada vez su eficacia respecto al tratamiento estándar “con criterios estrictos”.
Una mejora limitada
Algunos principios activos más modernos pasaron también los controles de las agencias de medicamentos y fueron tumbados por la farmacovigilancia. Ese es el caso del rimonabant, indicado para tratar la obesidad junto a la dieta y el ejercicio. La Unión Europea aprobó su salida al mercado en 2006 con el nombre de Acomplia. El laboratorio Sanofi-Aventis, propietario de la patente, intentó igualmente comercializarlo en Estados Unidos. La autoridad de regulación del país, la Food and Drug Administration (FDA), consideró que los datos de seguridad y de eficacia no eran suficientes. En 2007 se negó a autorizar este medicamento que prometía revolucionar el mercado de productos para adelgazar. Un par de años después, la EMA retiró el rimonabant del mercado europeo por provocar trastornos psicológicos, intentos de suicidio, o suicidios a secas.
Actualmente, un medicamento autorizado por la Agencia Europea de Medicamentos (EMA) -y luego por la Comisión Europea- tiene que pasar una segunda aprobación cinco años después de la primera. A partir de entonces, salvo que se hayan tomado medidas excepcionales de seguridad, la autorización de comercialización “será válida durante un periodo ilimitado”, explica la EMA en su web. La única barrera de seguridad que sigue a partir de este momento es la farmacovigilancia. Los medicamentos antiguos ya tienen esta autorización ilimitada.
“El sistema de regulación europeo es el más garantista del mundo”, asegura Emili Esteve, Director técnico de la asociación de farmacéuticas FarmaIndustria desde hace 15 años y ex empleado de la AEMPS [Nota de los editores de SyF: esta es la visión de la industria, la de los científicos independientes es otra: véase en esta misma sección la investigación sobre la vacuna para el Virus del Papiloma Humano]. Las agencias europeas forman parte del proyecto de armonización de los requisitos para evaluar los medicamentos lanzado en 1990 por el Consejo Internacional para la Armonización. Esta organización, que reúne reguladores e industrias farmacéuticas, creó un Documento Técnico Común que lista las informaciones que considera necesarias para medir la calidad, eficacia y la seguridad de un fármaco. Y, con Japón, la Unión Europea fue la única institución que volvió este formato obligatorio para las solicitudes de autorización de medicinas. El abogado especializado en temas de salud Francisco Almodóvar, que representa a mujeres españolas que tomaron Agreal, considera que “en Europa hay una red de farmacovigilancia de las más avanzadas del mundo”.
A pesar de todo, cuando Francia retiró la tirotricina de su mercado en 2005 por favorecer infecciones resistentes a los antibióticos, ningún país siguió su ejemplo. Tras la prohibición de benzbromarona por Portugal y Francia en 2003, España la ha seguido comercializando. En Países Bajos y Reino Unido se venden todavía medicamentos con pergolida, un principio activo que Estados Unidos y Canadá dejaron de comercializar en 2007 por causar valvulopatías.
Por el contrario, estos dos últimos Estados todavía autorizan la rosiglitazona, mientras que la Comisión Europea suspendió su autorización en 2010 por aumentar los “riesgos de problemas del corazón”. Y tanto la agencia estadounidense, la FDA, como la Europea, la EMA, consideran desde aquel mismo año que los riesgos de ataque del corazón y de apoplejía conllevados por la sibutramina no son compensados por su capacidad adelgazante. Ésta sigue sin embargo a la venta en Brasil y República Dominicana.
Para Francisco Almodóvar, la solución es la misma que la que proponía el economista Germán Velásquez: “el sistema de farmacovigilancia debería de ser único a nivel mundial”. Una medida que sería “positiva”, declara Emili Esteve, de FarmaIndustria, pero que, de momento, le resulta “utópica”.
Para ver el detalle de los diez casos, ir a:http://www.elmundo.es/grafico/salud/2017/01/16/57c99d8c22601d187d8b458c.html
|
“HOY EN DÍA LAS PRIORIDADES LAS MARCA EL QUE PONE EL DINERO” Germán Velásquez dimitió de su puesto de Director del Secretariado de la Salud Pública, de la Innovación y de la Propiedad Intelectual de la OMS en 2010. Uno de los motivos que le llevó a despedirse fue lo que llama “la privatización” de la institución. La OMS se financia de dos maneras: vía “contribuciones valoradas”, que son cuotas obligatorias pagadas por cada Estado miembro, y vía “contribuciones voluntarias” de origen público o privado. Las segundas han superado las primeras con creces. La Organización informa en su web oficial de que “estos últimos años, las contribuciones voluntarias han representado más de las tres cuartas partes de la financiación”. “El carácter voluntario de las contribuciones significa que [quienes las hacen] las pueden dar para determinados proyectos”, subraya Germán. Las contribuciones obligatorias de los Estados representan el 21% del presupuesto total de la OMS para 2016 y 2017, según el resumen de sus financiaciones que publica la Organización. Y de este dinero, casi un tercio se destina a servicios empresariales y a gastos de funcionamiento. Al final, el 82% de los gastos de la OMS en proyectos viene de contribuciones voluntarias. “Hoy en día las prioridades las pone el que pone el dinero”, analiza el economista colombiano. Para ver el financiamiento de la OMS ir a http://extranet.who.int/programmebudget/Biennium2016/Financing |