Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Ética, Derecho y Ensayos Clínicos
Adulteraciones y Falsificaciones
Bruselas solo prevé 16 excepciones a los mecanismos anti-falsificación
Carlos B Rodríguez
El Global, 11 de septiembre de 2015
http://www.elglobal.net/noticias-medicamento/2015-09-11/politica-sanitaria/bruselas-solo-preve-16-excepciones-a-los-mecanismos-anti-falsificacion/pagina.aspx?idart=930847&utm_source=mail&utm_medium=newsletter&utm_campaign=elglobal
El borrador de actos delegados de la Comisión Europea libra a la distribución de chequeos sistemáticos
Para finales del año 2018, como muy tarde, los fabricantes y re-envasadores de medicamentos que operan en el mercado farmacéutico europeo deberán haber implementado medidas de seguridad en la gran mayoría de los fármacos. Según el esperado borrador de los actos delegados a la directiva anti-falsificación, que la Comisión Europea publicó el pasado 12 de agosto, solo 14 productos o categorías de productos sujetos a receta (ver tabla) no tendrán que verse sometidos a estas medidas de seguridad. Por contra, de momento solo dos fármacos que no necesitan receta médica sí estarán sujetos a dichas medidas, ante el riesgo que presentan de ser falsificados.
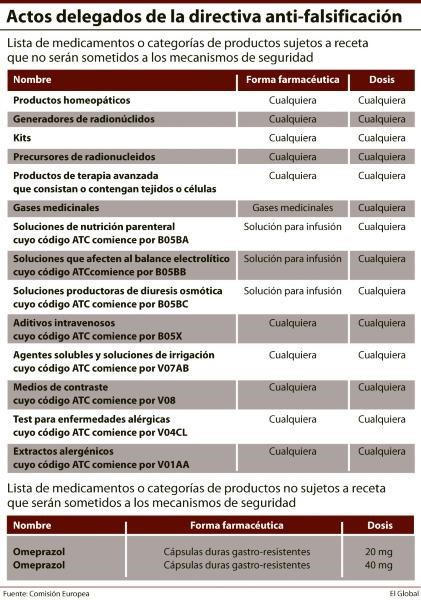
Ninguna de estas dos listas está cerrada. El borrador de los actos delegados ha establecido el mecanismo que permitirá a las autoridades nacionales competentes notificar a la Comisión acerca de todo producto no sujeto a receta médica que pueda estar en riesgo de falsificación, tan pronto como sean conscientes de que dicho riesgo existe; así como de todo fármaco sujeto a receta que no parezca estar en riesgo de ser pirateado. Dicha notificación deberá ir acompañada de una evaluación de los riesgos y de la documentación que acredite esa situación. Si, a la luz del número de muertes u hospitalizaciones debido a la exposición a las falsificaciones, la Comisión o cualquier estado considera que es necesaria una acción rápida, Bruselas dará una respuesta en un plazo máximo de 45 días.
El borrador invita a debatir acerca de cómo prevenir la entrada de medicamentos falsos en la cadena de suministro y está llamado a colocar la última piedra del proceso iniciado hace años para garantizar la seguridad de los pacientes como punto final de la cadena de suministro farmacéutico. Para ello requiere de manera obligatoria dos medidas clave: identificadores únicos y mecanismos anti-falsificación. Los aspectos técnicos presentados en el documento no establecen las características técnicas del segundo mecanismo anti-manipulación ya que el mandato de la Comisión solo cubría las características técnicas del identificador único.
Las claves del borrador
Junto al borrador de actos delegados, la directiva anti-falsificación también requirió de la Comisión Europea que llevara a cabo un estudio de evaluación de los beneficios, costes y coste-efectividad de las opciones técnicas para el identificador único (por ejemplo, ¿cuál será su composición y formato del código de barras?); las opciones para el grado de verificación de la autenticidad del medicamento, especificando también las medidas de seguridad y los acuerdos prácticos para tal verificación (¿Quién chequeará las medidas de seguridad y cuándo?) y las opciones técnicas para establecer y gestionar el sistema repositorio (¿Quién lo establecerá? ¿Quién lo gestionará? ¿Quién lo supervisará?)
La primera conclusión de este estudio señala que la composición y el formato del identificador único deberían estar plenamente armonizado a nivel europeo. El identificador debería estar incluido en un código de barras 2D y contener el código del producto, un número de serie, el número de reembolso nacional, el número del lote y la fecha de caducidad.
Asimismo, el estudio señala que la autenticidad de los medicamentos debería estar garantizada por un sistema de verificación de principio a fin complementado por comprobaciones puntuales por parte de los distribuidores. Es decir, que los medicamentos deberán ser sistemáticamente verificados por las farmacias antes de ser dispensados al público (la puerta queda abierta en el caso de las farmacias de hospital, al dejar a las instituciones decidir en qué parte del proceso llevar a cabo la verificación), y sólo aquellos productos con mayor riesgo de ser falsificados deberían ser chequeadas de forma adicional por la distribución.
En cuanto a los sistemas repositorios, la Comisión Europea cree que deben estar físicamente localizados en la Unión Europea y ser establecidos y gestionados por los agentes. Las autoridades nacionales competetentes, no obstante, deberían ser capaces de acceder a los mismos y supervisarlos.
Finalmente el borrador prevé la adopción de medidas para garantizar, tal y como establece la normativa comunitaria, la protección de los datos personales, los intereses legítimos para proteger información comercial de carácter confidencial y la propiedad y confidencialidad de los datos generados por el uso de los mecanismos de seguridad. La Comisión resalta que la regulación delegada no requiere que ningún dato personal sea almacenado en los sistemas repositorios. Las medidas son más bien preventivas, señala, por lo que garantizar la protección de los datos personales en el caso de los usuarios de los repositorios (por ejemplo, los farmacéuticos) implicaría decidir utilizar el repositorio para fines ajenos al ámbito de aplicación del Reglamento Delegado, e implicar el uso / almacenamiento / manipulación de los datos de los pacientes en los repositorios (por ejemplo: e- recetas ) .
Futuros pasos
Todavía cabe esperar cambios en el borrador. Los agentes interesados pueden enviar sus comentarios hasta el 11 de octubre. En casi todos los países, los actos delegados tendrán que estar implementados para finales de 2018. Los fármacos puestos a la venta antes de la fecha de aplicación de la Regulación podrán continuar dispensándose hasta su fecha de caducidad. Tres países, no obstante, tienen una moratoria de seis años. Cuando la directiva anti-falsificación entró en vigor, Bélgica, Grecia e Italia ya aplicaban sistemas para verificar la autenticidad de los medicamentos. El borrador les concede por lo tanto un plazo de nueve años desde la publicación de los actos delegados para adaptar sus respectivos sistemas y armonizarlos con los de la Unión.